
Nephrolithiasis and/or nephrocalcinosis (management and evaluation)
exp date isn't null, but text field is
- Nephrocalcinosis (NC) describes the deposition of calcium salts in the tubules, tubular epithelium and/or the interstitial tissue of the kidney. It may be detected incidentally (e.g. ultrasound for another indication) without evidence of kidney dysfunction, or it may result in acute or chronic kidney injury. Ultrasound and CT are the most sensitive tests for the detection of NC, whereas MRI is not ideal for visualising calcification. Imaging studies are unable to distinguish the composition of such calcium deposits but biochemical analysis invariably shows evidence of either calcium phosphate or calcium oxalate (sometimes referred to as “oxalosis”) deposition. NC may be categorised as medullary, cortical or diffuse according to the location of such deposition. Medullary NC is by far the most common form.
- Urolithiasis (UL) is the presence of stones/calculi anywhere within the urinary tract and the term nephrolithiasis refers to stones localised within the kidney. UL is much less common in children than adults, but the incidence is rising rapidly in the developed world (1). Children are more likely to have an underlying risk factor, such as a metabolic predisposition for stone formation, compared with adults in whom the majority of cases are idiopathic (2). Aside from a predisposing condition, factors including diet (high protein and sodium intake) and increased rates of obesity have been implicated with this rising incidence. Despite this, children with urolithiasis are more likely to have a below average weight at the time of presentation (2). Boys are more likely to develop UL than girls in the first decade of life although this is reversed from the second decade onwards (3).
There is considerable overlap in the causes of NC and UL. NC is a risk factor for urolithiasis (UL) but does not necessarily lead to the development of stones.
- Hypercalciuria (which may occur in the context of hypercalcaemia or normocalcaemia):
- Idiopathic hypercalciuria – the most common cause of hypercalciuria
- Increased sodium/salt intake (leads to increased distal tubular secretion of calcium)
- Vitamin A, C and D excess or intoxication
- Prolonged immobility due to increased bone resorption leading to resorptive hypercalciuria
- Hyperparathyroidism – may be primary, secondary or tertiary
- Hypophosphatasia
- Hypophosphataemia
- Ketogenic diet
- Malignancy with paraneoplastic effects
- Sarcoidosis and other granulomatous diseases
- Milk-alkali syndrome – hypercalcaemia and metabolic alkalosis secondary to high intake of calcium (usually dietary supplements) and absorbable alkali (often in the form of antacid medications)
- Inherited disorders affecting the renal tubules (see below)
- Medications:
- Loop diuretics (e.g. furosemide) and some potassium-sparing diuretics (e.g. triameterene)
- Glucocorticoids (e.g. dexamethasone)
- Acetazolamide and other carbonic anhydrase inhibitors
- Topiramate
- Calcium supplementation
- Phosphate-containing laxatives (when taken in significant quantity such as in laxative abuse)
- Magnesium trisilicate
- Vitamins A, C and D (e.g. intoxication from parenteral nutrition)
- Ciprofloxacin
- Ceftriaxone
- Aciclovir
- Sulfa-containing medications
- Ethylene glycol – directly converted to glycolate and oxalate
- Protease inhibitors (e.g. indinavir and lopinavir)
- Probenecid
- Prematurity (particularly related to furosemide exposure)
- Inherited disorders:
- Distal / type 1 renal tubular acidosis (RTA) – a group of disorders characterised by impaired tubular H+ excretion leading to metabolic acidosis, elevated urine pH (> 5.5), hypocitraturia, hypercalciuria, hypokalaemia, and failure to thrive. Patients may also have sensorineural hearing loss.
- Bartter syndrome – a group of disorders characterised by impaired sodium, chloride and potassium reabsorption in the thick ascending limb of the loop of Henle. This leads to reduced calcium reabsorption and subsequent hypercalciuria. The biochemical disturbance is similar to that seen with loop diuretic therapy. The more severe antenatal/neonatal form is caused by variants in the SLC12A1 gene affecting the Na-K-Cl co-transporter.
- Familial hypomagnesaemia with hypercalciuria and nephrocalcinosis (FHHNC) – autosomal recessive condition associated with impaired calcium and magnesium reabsorption in the thick ascending limb of the loop of Henle. Variants occur in the CLDN16 or CLDN19 genes encoding the tight junction proteins claudin 16 and 19. Patients with variants in CLDN19 also present with ocular abnormalities including myopia and nystagmus.
- Dent’s disease – a group of X-linked recessive conditions. The majority of cases are secondary to variants in the CLCN5 gene, which encodes a proximal tubular chloride channel. This leads to low molecular weight proteinuria and hypercalciuria, with the development of NC and progressive deterioration in kidney function over time.
- Cystinosis – an autosomal recessive lysosomal storage disorder due to variants in the CTNS gene encoding the protein cystinosin. This is associated with accumulation of the amino acid cystine. Patients develop proximal / type 2 RTA (renal Fanconi syndrome), hypercalciuria/NC, progressive renal impairment, growth failure and eye problems due to systemic cystine deposition.
- Lowe’s syndrome (oculocerebrorenal syndrome) – an X-linked recessive disorder due to variants in the OCRL1 The phenotype includes cataracts, hypotonia, developmental delay and type 2 RTA with hypercalciuria/NC.
- Wilson’s disease – an autosomal recessive disorder associated with impaired copper excretion. It may lead to type 2 RTA with associated hypercalciuria/NC.
- Tyrosinaemia – a group of autosomal recessive disorders associated with inability to break down the amino acid tyrosine. Patients may also develop a type 2 RTA with hypercalciuria/NC.
- Enamel-renal syndrome – autosomal recessive disorder due to variants in the FAM20A Patients have hypocalcaemia but present with NC.
- William’s syndrome – caused by chromosome 7 microdeletion. This predisposes to hypercalcaemia (cause not completely understood) and therefore hypercalciuria/NC.
- Hereditary hypophosphataemic rickets with hypercalciuria (HHRH) – an autosomal recessive condition due to variants in the NPT2C gene encoding the sodium-phosphate co-transporter. This results in urinary phosphate wasting, hypophosphataemic rickets, and inappropriately elevated 1,25(OH)2D (calcitriol) levels leading to hypercalciuria/NC.
- CYP24A1 gene variants – this gene encodes a member of the cytochrome P450 superfamily of enzymes. The protein product (CYP24A1) helps catalyse the degradation of 1,25(OH)2 Variants in CYP24A1 may lead to hypervitaminosis D and associated hypercalciuria/NC (particularly infantile hypercalcaemia/hypercalciuria).
- Sickle cell disease
- Metabolic disorders and abnormalities:
- Hyperoxaluria:
- Primary hyperoxaluria (PH types 1, 2 or 3) – a group of rare autosomal recessive conditions associated with excess oxalate production and elevated urine oxalate levels. PH type 1 (PH1) is the most severe phenotypic form. It has an estimated incidence of 1:100,000 and is caused by variants in the AGXT Patients typically present in childhood with NC, recurrent UL, and progressive renal failure. Serum oxalate levels typically remain normal (i.e. < 10 µmol/L) until they reach stage 4 chronic kidney disease (CKD). In PH1, other organs may also be involved including the bones (pain and anaemia), eyes (retinopathy), nerves (neuropathy) and heart (arrhythmia and heart block). PH2 is rarer still and patients may present around adolescence with oxalate stones. CKD develops in ~10% of PH2 patients. PH3 has the mildest phenotype. For all patient with PH, dietary modification is not helpful as oxalate production is a generated by metabolic pathways rather than from an external source.
- Secondary (enteric) hyperoxaluria – due to increased enteric oxalate absorption. This is associated with malabsorptive states such as short gut syndrome, inflammatory bowel disease and cystic fibrosis. Fat malabsorption causes increased binding of free fatty acids to dietary calcium. This thereby reduces the amount of calcium in the gut which would normally help to precipitate dietary oxalate. Furthermore, increased delivery of unabsorbed fatty acids and bile salts to the large intestine increases colonic permeability and leads to hyper-absorption of oxalate thus raising serum oxalate levels and predisposing to the formation of oxalate-containing stones.
- Idiopathic hyperoxaluria - high dietary consumption of oxalate (as seen in vegetarians) or low calcium intake may be predisposing factors to stone formation.
- Cystinuria – an inherited disorder with an estimated incidence of 1:10,000. It is an autosomal recessive condition caused by variants in the SLC3A1 or SLC7A9 genes respectively. It is associated with impaired tubular reabsorption of the dibasic amino acids cystine, ornithine, lysine and arginine (‘COLA’). Only cystine excretion is clinically relevant as it readily precipitates and may lead to stone formation. Cystinuria is responsible for approximately 5-10% of childhood UL and it is often associated with the formation of bladder stones. Cystine crystals have a characteristic hexagonal shape on microscopy. At a urinary pH of 5-7, cystine is poorly soluble and more likely to precipitate into stones.
- Hypocitraturia – citrate is a natural inhibitor of calcium phosphate and calcium oxalate formation within the urinary tract. It binds with calcium to form a soluble complex that then reduces the amount of free calcium in the urine. The lower the free calcium concentration, the less calcium is available to bind with oxalate, which is a risk factor for stone formation. Hypocitraturia may be seen in around 10% of children with UL (3). Chronic acidosis may lead to hypocitraturia due to increased proximal tubular reabsorption of citrate.
- Disorders of purine metabolism (predispose to the formation of purine stones including urate stones):
- Hyperuricaemia/hyperuricosuria secondary to chemotherapy (tumour lysis syndrome) and myelodysplastic syndrome (rare in children).
- Lesch-Nyhan syndrome (HGPRT deficiency) – an autosomal recessive disorder associated with hyperuricaemia/hyperuricosuria, formation of urate stones, abnormal choreoathetoid movements, self-mutilating behaviours (e.g. tongue biting), developmental delay, and progressive renal impairment.
- Adenine phosphoribosyltransferase (APRT) deficiency – an autosomal recessive disorder associated with build-up of 2,8-dihydroxyadenine (rather than adenine) and the formation of stones which are similarly radiolucent and have identical chemical reactivity to urate stones.
- Hereditary xanthinuria – an autosomal recessive disorder due to xanthine oxidase (XO) deficiency. XO converts xanthine to urate so xanthine build-up occurs leading to the formation of xanthine stones and hypouricaemia/hypouricosuria.
- Renal hypouricaemia – a disorder due to loss of function variants in the genes encoding the renal urate transporters (e.g. SLC22A12 encoding URAT1, and SLC2A9 encoding GLUT9). These variants reduce tubular urate reabsorption and lead to hypouricaemia/hyperuricosuria.
- Hyperoxaluria:
- Endocrine disorders:
- Hypo- and hyperthyroidism
- Cushing syndrome – can cause hypercalciuria
- Adrenal insufficiency
- Other conditions:
- Medullary sponge kidney (tubular ectasia)
- Fat necrosis
- Acute cortical necrosis (e.g. preterm infants with prolonged ischaemic down-time at birth) – these patients may have evidence of cortical (rather than medullary) NC.
- A metabolic predisposition is identified in up to 50% of children with UL and may be due to:
- Increased urinary excretion of ions/metabolites such as calcium, phosphate, oxalate and cystine, leading to urinary solute supersaturation.
- Altered urinary pH affecting the solubility of these ions/metabolites, thus affecting urinary excretion.
- A reduction in natural inhibitors of urinary stone formation (particularly citrate).
- An infective predisposition/cause is identified in up to 25% of children and may be due to:
- Urinary stasis e.g. due to poor bladder emptying or anatomical abnormalities including pelviureteric junction (PUJ) obstruction, horseshoe kidney and duplex collecting system. Urinary diversion techniques and bladder augmentation (cystoplasty) are also associated with increased risk of UL primarily due to risk of urinary stasis and infection.
- Certain bacteria (e.g. Proteus, Providencia, Klebsiella, Pseudomonas, Enterobacter) produce high levels of urease which convert urea to ammonium and bicarbonate, thus making the urine more alkaline. A persistent urine pH > 7 predisposes to the formation of struvite and calcium phosphate stones. Struvite stones are more likely to fill the renal calyces producing a typical ‘staghorn’ appearance.
- Other stones may be idiopathic in nature with no identified cause (more common in adults).
Stone composition is determined by the underlying cause for stone formation.
Case series have demonstrated the following data on stone composition and frequency in children (4, 5):
- Calcium oxalate stones – 45-65%
- Calcium phosphate stones – 14-30%
- Struvite (magnesium ammonium phosphate) stones – 13%
- Cystine stones – 5%
- Urate stones – 4%
- Mixed or miscellaneous (including xanthine) stones – 4%
Stones < 5 mm can usually pass through the urinary tract spontaneously. Those 5-7 mm in size have a 50% likelihood of spontaneous passage. Those > 7 mm are unlikely to pass without urological intervention.
- Nausea ± vomiting
- Frank or microscopic haematuria
- Passage of a stone or gravel-like sediment
- Abdominal or flank pain which is typically unilateral. This may vary from a diffuse and dull ache to severe colicky pain. Note that classic renal colic may be absent in young children <5 years of age.
- Pain may be referred to the ipsilateral testicle and tip of penis in boys, or labia in girls
- Strangury (slow, painful and hesitant urination) which may be associated with bladder stones obstructing the bladder outlet
- Fever and dysuria associated with UTI
- Acute kidney injury
- Physical examination may reveal costovertebral angle tenderness
- Stones are often found incidentally without any clinical features
- Detailed birth history including prematurity and medications which may been given in the neonatal period
- Dietary history
- Drug history (including vitamin supplementation)
- Family history of NC/UL, renal impairment (and need for dialysis or kidney transplant), hearing problems (e.g. dRTA and Bartter), eye problems (e.g. FHHNC)
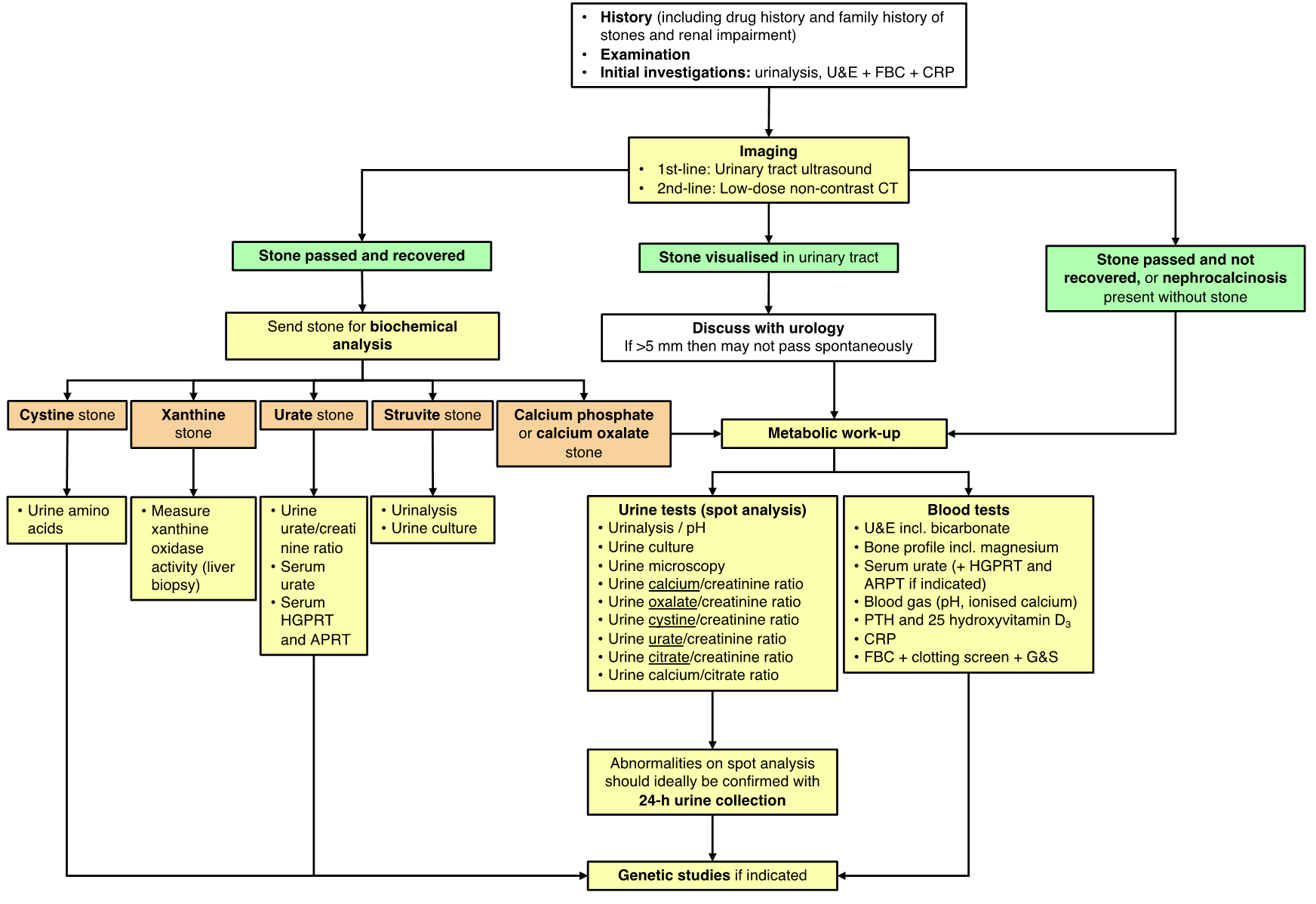
See Appendix I for a diagnostic pathway for investigating UL.
- Imaging:
- Urinary tract ultrasound is the first-line imaging modality in children when UL is suspected. This should include visualisation of the kidney, a fluid-filled bladder and the ureters adjacent to the kidney and the bladder.
- Low-dose non-contrast CT can be used as a second-line investigation if ultrasound is inconclusive.
- X-ray KUB may show up radiopaque stones if > 3 mm in size (e.g. calcium oxalate, calcium phosphate, struvite). It will not show radiolucent stones (e.g. urate stones) and cannot demonstrate obstruction. However it may useful to demonstrate stone burden and is a useful adjunct in planning surgery.
- DMSA scan may be used to evaluate renal function if there is concern regarding longstanding obstruction.
- Biochemical analysis of a stone (if available) or urinary sediment/gravel may help indicate the cause for stone formation.
- Urine tests:
- Urinalysis for red cells, white cells, leucocytes and nitrites and urine pH
- Urine microscopy for assessment of crystals
- Urine culture to assess for infection
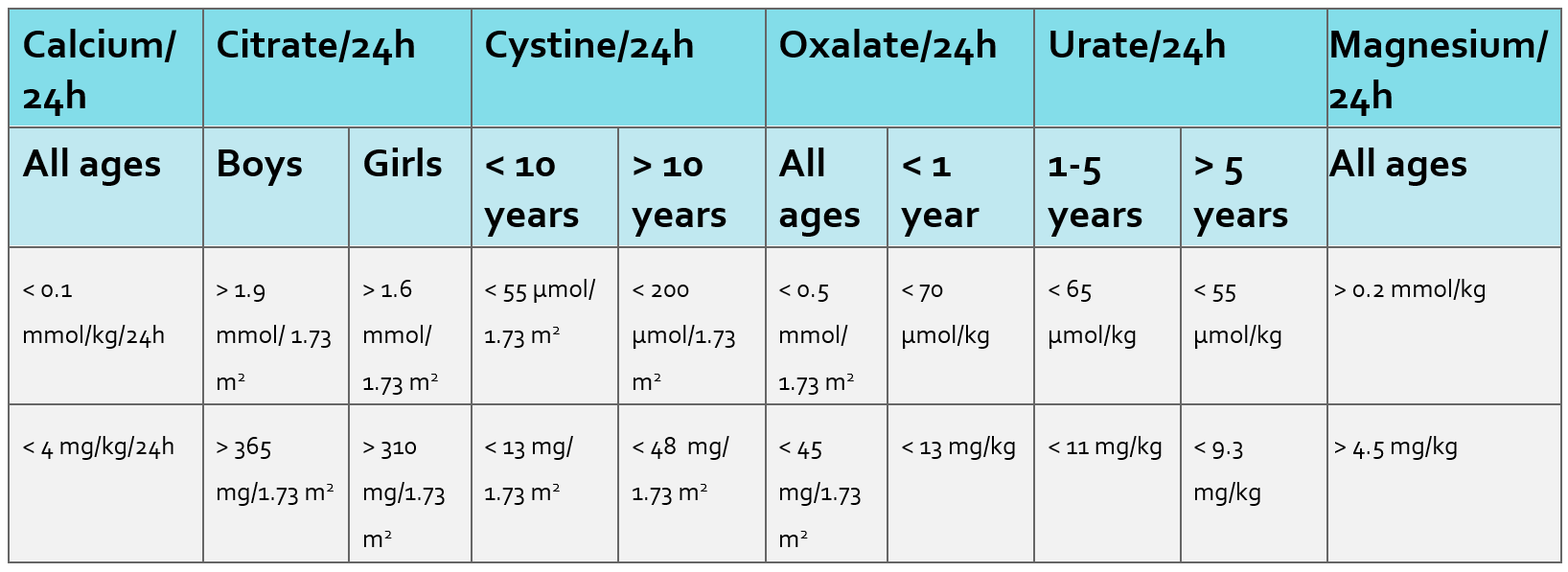
- Urine biochemistry which can be sent as a spot/random sample (see Appendix II/III for reference values) and followed up with a 24-hour collection (see Appendix IV for reference values) if abnormalities are detected:
- Urine calcium (including calcium/creatinine ratio)
- Urine oxalate (including oxalate/creatinine ratio)
- Urine cystine (including cystine/creatinine ratio)
- Urine urate (including urate/creatinine ratio)
- Urine citrate (including calcium/citrate ratio)
- Urine creatinine (essential if sending the above tests to allow for ratio calculation and in a 24-hour sample this allows for assessment of adequacy of collection [girls: 130-170 µmol/kg/day and boys 170-210 µmol/kg/day]).
- Urine magnesium – not routine
- Urine phosphate – not routine
- Urine amino acids if cystinuria is suspected. The cyanide-nitroprusside test (CNT) is a rapid screening test for cystinuria but it has low sensitivity and its use is not routinely recommended (6).
- Blood tests:
- Creatinine, urea, sodium, potassium, chloride
- Magnesium, phosphate, calcium (including ionised calcium), albumin
- Urate and specific metabolic markers for purine disorders if indicated (e.g. APRT and HGPRT)
- Vitamin D (25(OH)D3) level should be considered
- Calcitriol level (1,25(OH)2D) may be of use if there is a suspected CYP24A1 variant
- Blood gas for pH, pCO2, HCO3-, base excess and ionised calcium
- FBC and CRP if infection suspected
- Coagulation screen and group and save if surgical intervention likely to be required
- Genetic testing can be sent if there is a suspected inherited cause for NC/UL. A monogenic cause should be suspected if there is early onset of stone formation, family history, parental consanguinity, highly-active stone disease affecting both kidneys and with large stone burden, underlying NC or renal hyperechogenicity in addition to UL, tubular dysfunction, evidence of CKD, extra-renal manifestation (e.g. hearing loss, eye problems, neurological problems, bone disease), and particular stone composition (e.g. cystine, xanthine). A NC genetic panel is currently offered by Dundee.
The acute management of urolithiasis is outlined by the NICE guidelines (7). A summary of their recommendations is listed below. If a stone is identified on imaging then discussion with the urology team is advised.
- Pain management:
- A non-steroidal anti-inflammatory drug (NSAID) by any route should be used as first-line analgesia (provided no known renal dysfunction)
- If NSAIDs do not offer significant pain relief, or are contraindicated, then give paracetamol and consider giving this parenterally rather than orally
- Offer opiates as a third-line option if NSAIDs and paracetamol are insufficient
- Start parenteral antibiotics early if suspected UTI or likely to be going for surgical intervention.
- Watchful waiting may be considered if the stone is < 5 mm in size.
- For distal ureteral stones < 10 mm in size, alpha-blockers (e.g. tamsulosin) may be considered along with hyperhydration. Tamsulosin dose is 0.2 mg OD (< 4 years), or 0.4 mg OD (> 4 years).
- Obstructing stones are an emergency and require urgent discussion with urology due to risk of parenchymal damage to the kidney. Surgical intervention may be required (e.g. with percutaneous drainage or ureteral stenting). Early surgical intervention is considered in cases with sepsis, renal impairment, solitary kidney, or gross hydronephrosis.
- If septic then definitive surgical intervention may be deferred until this has been managed with antibiotics and/or drainage due to risk of further septic shower when stones are mobilised or broken down.
- The European Association of Urology (EAU) recommends the following surgical options for children (8):
- Ureteral stones– can be managed with shock wave lithotripsy (SWL), if localisation is possible, or by ureteroscopy (URS).
- Pelvic stones < 20 mm in size – SWL can be offered in children.
- Pelvic stones > 20 mm in size – percutaneous nephrolithotomy (PCNL) can be offered, including for staghorn stones.
Medical management is guided by the underlying cause (summarised in Appendix V).
- Any causative agent (e.g. furosemide) should be stopped if possible.
- Liberal fluid intake (>1.5 L/m2/day) is likely to be of benefit in helping reduce the risk of stone formation.
- Dietary advice for those with hypercalciuria and/or hypocitraturia:
- Adding fresh lemon juice to water as a source of citrate
- Avoiding carbonated drinks
- Salt restriction of 2-6 g/day depending on age
- Maintaining normal calcium intake of between 350 mg/day for children and 1000 mg/day for adolescents
- Dietary restriction of animal protein (although this remains controversial in children and is not routinely advised)
- Dietary advice for those with cystinuria:
- Salt restriction
- Reduced methionine intake (lowers cystine production). Foods high in methionine include certain fish (dried cod, tuna, sardines), shellfish (lobster, crayfish), liver, poultry, beef, cheese, and eggs.
- Medications may be indicated depending on the underlying aetiology:
- Thiazide diuretics (e.g. chlorothiazide, hydrochlorothiazide, bendroflumethiazide) – should be considered if hypercalciuria is present without Volume depletion stimulated by thiazides promotes both sodium and calcium reabsorption in the proximal tubule, thereby reducing urinary calcium excretion. Thiazides may induce hypokalaemia so serum potassium should be monitored after starting, or after any dose increment. Thiazides are useful alongside dietary sodium restriction.
- Chlorothiazide dose is 10-20 mg/kg BD.
- Citrate (typically given in the form of potassium citrate) – should be considered for those with recurrent stones that are > 50% calcium oxalate (i.e. those with hyperoxaluria), and for those with hypercalciuria, hypocitraturia and cystinuria. Citrate binds intestinal and urine calcium and increases urine pH. It is converted to bicarbonate in the liver and also works as a urinary alkalinising agent. The increased urinary pH allows for increased urinary calcium solubility, thus aiding excretion. In hypercalciuria, excessive alkalinisation of the urine (urine pH >7.5) is a risk factor for phosphate precipitation so the dose of citrate may need to be titrated once urine pH exceeds this threshold. In cystinuria, the solubility of cystine increases threefold with a urinary pH > 8 so this is an important therapeutic agent in helping reduce stone formation (aim to reduce urine cystine levels to < 1 mmol/L). Realistically, urine pH should be maintained between 7.5 and 8 in cystinuria. Serum potassium and bicarbonate levels should be monitored whilst on potassium citrate treatment.
- Potassium citrate starting dose is 0.5-1.0 mmol/kg/day (0.1-0.2 g/kg/day) usually given in 3 divided doses/day.
- Potassium supplementation (on its own without citrate) will inhibit distal sodium reabsorption and this acts to reduce urine calcium excretion.
- Pyridoxine (vitamin B6) – can be used in high doses (2-5 mg/kg, and increased up to 20 mg/kg) for those with PH1 and a minority of patients may be able to achieve normal urine oxalate levels with this treatment, although studies report variable results (9). Pyridoxine works as a co-factor of AGTX and pyridoxine deficiency is a known risk factor for oxaluria.
- Chelating agents (e.g. penicillamine or tiopronin [Thiola®]) – may be used in cystinuria where there is refractory disease despite conservative treatment and urinary alkalinisation. Chelating agents cleave the disulphide bond of the cystine molecule, releasing 2 cysteine moieties. Penicillamine and tiopronin can both then bind to cysteine to form more soluble complexes which are more readily excreted in the urine. Both drugs have similar and potentially serious side effects that require close monitoring (i.e. regular urinalysis and FBC monitoring). These include proteinuria, haematuria, thrombocytopenia, anaemia, alopecia, connective tissue disorders and pyridoxine deficiency (pyridoxine supplementation may therefore be required).
- Penicillamine dose is 20-30 mg/kg given in 4 divided doses/day, and the dose of tiopronin is 15-40 mg/kg/day given in 3 divided doses/day (adult dose is 800-1500 mg/day in 3 divided doses/day) (6).
- Uric acid lowering therapies may be used in those with hyperuricaemia/hyperuricosuria. These include xanthine oxidase inhibitors (e.g. allopurinol and febuxostat) and recombinant urate oxidases (e.g. rasburicase and pegloticase). The latter are used for acute urate lowering as in tumour lysis syndrome.
- Allopurinol dose in children is 10-20 mg/kg daily, up to a maximum of 400 mg/day.
- Thiazide diuretics (e.g. chlorothiazide, hydrochlorothiazide, bendroflumethiazide) – should be considered if hypercalciuria is present without Volume depletion stimulated by thiazides promotes both sodium and calcium reabsorption in the proximal tubule, thereby reducing urinary calcium excretion. Thiazides may induce hypokalaemia so serum potassium should be monitored after starting, or after any dose increment. Thiazides are useful alongside dietary sodium restriction.
The prognosis is also guided by the underlying cause. NC may be reversed, particularly where a causative agent (such as furosemide) is stopped. In most cases, NC is not associated with kidney dysfunction. However, the formation of UL may cause obstruction which can then contribute to acute kidney injury. Furthermore, underlying genetic disorders such as Dent’s disease, FHHNC, and PH may all be associated with the development of chronic kidney disease potentially leading to end stage kidney disease (ESKD) and the need for renal replacement therapy (RRT). Of these 3 conditions, PH is the most likely to lead to ESKD in childhood or adolescence.
Adapted from Hulton, Arch Dis Child, 2001 (10).
|
Parameter and patient age |
Ratio of solute to creatinine |
Units |
|
Calcium |
mmol/mmol |
mg/mg |
|
< 12 months |
< 2.2 |
< 0.78-0.81 |
|
1-3 years |
< 1.5 |
< 0.50-0.53 |
|
3-5 years |
< 1.1 |
< 0.39-0.41 |
|
5-7 years |
< 0.8 |
< 0.28-0.30 |
|
> 7 years |
< 0.7 |
< 0.21-0.24 |
|
Oxalate |
mmol/mol |
mg/g |
|
0-6 months |
< 325-360 |
< 260-288 |
|
7-24 months |
< 132-174 |
< 110-139 |
|
2-5 years |
< 98-101 |
< 80 |
|
6-14 years |
< 70-82 |
< 60-65 |
|
> 16 years |
< 40 |
< 32 |
|
Cystine |
mmol/mol |
mg/g |
|
< 1 month |
< 85 |
< 180 |
|
1-6 months |
< 53 |
< 112 |
|
> 6 months |
< 18 |
< 38 |
|
Urate |
mmol/mmol (5th-95th percentile) |
mg/mg (5th-95th percentile) |
|
1-6 months |
0.80-1.60 |
1.189-2.378 |
|
7-12 months |
0.70-1.50 |
1.040-2.229 |
|
1-2 years |
0.50-1.40 |
0.743-2.080 |
|
2-3 years |
0.47-1.30 |
0.698-1.932 |
|
3-5 years |
0.40-1.10 |
0.594-1.635 |
|
5-7 years |
0.20-0.80 |
0.446-1.189 |
|
7-10 years |
0.26-0.56 |
0.386-0.832 |
|
10-14 years |
0.20-0.44 |
0.297-0.654 |
|
14-17 years |
0.20-0.40 |
0.297-0.594 |
|
Citrate (hypocitraturia is risk factor for stone formation) |
mmol/mmol |
g/g |
|
0-5 years |
> 0.25 |
> 0.42 |
|
> 6 years |
> 0.15 |
> 0.25 |
|
Magnesium (hypomagnesuria is risk factor for stone formation) |
mmol/mmol |
g/g |
|
1-12 months |
> 2.20 |
> 0.48 |
|
1-2 years |
> 1.70 |
> 0.37 |
|
2-3 years |
> 1.60 |
> 0.34 |
|
3-5 years |
> 1.30 |
> 0.29 |
|
5-7 years |
> 1.00 |
> 0.21 |
|
7-10 years |
> 0.90 |
> 0.18 |
|
10-14 years |
> 0.70 |
> 0.15 |
|
14-17 years |
> 0.60 |
> 0.13 |
|
Patient age |
Ratio of calcium to citrate in 24-hour urine |
|||
|
mmol/mmol (5th-95th percentile) |
mg/mg (5th-95th percentile) |
|||
|
|
Boys |
Girls |
Boys |
Girls |
|
2-6 years |
0.24-2.30 |
0.14-2.0 |
0.05-0.48 |
0.03-0.42 |
|
7-12 years |
0.24-2.90 |
0.19-2.30 |
0.05-0.60 |
0.04-0.47 |
|
> 13 years |
0.29-3.80 |
0.24-2.90 |
0.06-0.80 |
0.05-0.60 |
|
Increased risk of stone formation with urine calcium/creatinine ≥ 1.6 mmol/mmol or ≥ 0.326 mg/mg |
||||

- VanDervoort K, Wiesen J, Frank R, Vento S, Crosby V, Chandra M, et al. Urolithiasis in pediatric patients: a single center study of incidence, clinical presentation and outcome. J Urol. 2007;177(6):2300-5.
- Issler N, Dufek S, Kleta R, Bockenhauer D, Smeulders N, Van't Hoff W. Epidemiology of paediatric renal stone disease: a 22-year single centre experience in the UK. BMC Nephrol. 2017;18(1):136.
- Novak TE, Lakshmanan Y, Trock BJ, Gearhart JP, Matlaga BR. Sex prevalence of pediatric kidney stone disease in the United States: an epidemiologic investigation. Urology. 2009;74(1):104-7.
- Miller LA, Stapleton FB. Urinary citrate excretion in children with hypercalciuria. J Pediatr. 1985;107(2):263-6.
- Milliner DS, Murphy ME. Urolithiasis in pediatric patients. Mayo Clin Proc. 1993;68(3):241-8.
- Stapleton FB. Childhood stones. Endocrinol Metab Clin North Am. 2002;31(4):1001-15, ix.
- Servais A, Thomas K, Dello Strologo L, Sayer JA, Bekri S, Bertholet-Thomas A, et al. Cystinuria: clinical practice recommendation. Kidney Int. 2020;S0085-2538(20):30829-2. Epub ahead of print.
- NICE Guideline [NG118]. Renal and ureteric stones: assessment and management. BJU Int. 2019;123(2):220-32.
- EAU Guidelines. Edn. presented at the EAU Annual Congress Amsterdam 2020. ISBN 978-94-92671-07-3.
- Ferraro PM, Taylor EN, Gambaro G, Curhan GC. Vitamin B6 intake and the risk of incident kidney stones. Urolithiasis. 2018;46(3):265-70.
- Hulton SA. Evaluation of urinary tract calculi in children. Arch Dis Child. 2001;84(4):320-3.
- Hoppe B, Kemper MJ. Diagnostic examination of the child with urolithiasis or nephrocalcinosis. Pediatr Nephrol. 2010;25(3):403-13.
Other useful resources
Rees L, Bockenhauer D, Webb NJA, Punaro MG. Paediatric Nephrology (Oxford Specialist Handbooks in Paediatrics). 3rd ed. Oxford: Oxford University Press; 2019.
Last reviewed: 17 November 2021
Next review: 30 November 2024
Author(s): Douglas Stewart, ST7 in Paediatric Nephrology
Approved By: Paediatrics Risk & Clinical Effectiveness Committee
Document Id: 973