
Renal anomalies detected or suspected antenatally
exp date isn't null, but text field is
Objectives
This guideline is intended to provide the following guidance:
- To outline which initial investigations are appropriate for congenital anomalies of the kidney and urinary tract that have been identified, or are suspected antenatally.
- To identify when onward referral to the nephrology/ urology/ genetics teams is appropriate.
- To provide a time frame over which the above should occur.
- To identify infants requiring antibiotic prophylaxis whilst awaiting investigation.
It is possible that infants with severe congenital renal anomalies have already been discussed antenatally within a multidisciplinary forum; such infants may already have plans for management in place that should be adhered to. Where infants are referred for imaging or radionuclide studies but do not yet require referral to specialist services, arrangements for follow up should be made as per local protocol.
Audience
This guideline is applicable to all medical, nursing and midwifery staff working with neonates in the West-of-Scotland managed clinical network.
Advice for parents
Advice for parents is also available online at infokid.org.uk This website contains information on a comprehensive range of congenital renal abnormalities.
Antibiotic prophylaxis
- Oral Trimethoprim – 2mg/kg daily, at night.
Renal abnormalities are the commonest congenital malformation with a mean prevalence of around 1.6 per 1000 births in one European study of over 709,030 births. The development of the kidney results from an interaction between the ureteral bud and the metanephrogenic mesoderm. At the 5th week of embryonic development the ureteral bud grows and comes into close contact with mesodermal elements. The ureteral bud undergoes a series of divisions which give rise to the pelvicalyceal system at the same time the mesoderm differentiates to form nephrons. The foetal kidneys can be easily visualised on ultrasound scan at 18 weeks gestation at which point it is possible to make measurements of the renal pelvic diameter. There is general consensus that a foetal renal pelvic diameter >10mm at 26 weeks gestation is indicative of significant dilatation.
The primary aim of postnatal investigation and management of these anomalies is to identify associated vesicoureteric reflux or indeed obstruction to the urinary tract and to minimise the risk of infection and resultant scarring of renal tissue. The long term aim is to preserve renal function.
With universal screening by antenatal ultrasound and improved scanning techniques, the rate of detection of congenital renal anomalies has increased over recent years. Consequently, a larger number of asymptomatic infants are being diagnosed prenatally. With increasing numbers of asymptomatic infants identified the trend has been to increase the threshold for intervention. However, there remains a requirement for targeted investigation in the immediate postnatal period to identify infants at risk of reflux, obstruction & infection.
Contents:
- Renal Pelvicalyceal Dilatation (RPD)
- Pelvi-Ureteric Junction (PUJ) Obstruction
- Vesico-Ureteric Junction (VUJ) Obstruction
- Ureterocoele
- Megaureter
- Posterior Urethral Valves
- Familial Vesicoureteric Reflux (VUR) – Screening siblings of affected children.
- Duplication of the Ureters (Duplex Collecting System/ Duplex Kidney(s)).
Renal Pelvicalyceal Dilatation (RPD)
|
Antenatal Counselling |
Consider neonatology counselling for low risk pathway (see flowchart) Neonatology counselling for high risk pathway (see flowchart) with Nephrology/Urology input. |
|
Attendance at Delivery/ Admission |
Not routinely required. NNU admission not routinely required. |
|
Imaging |
Low risk pathway - Renal USS 4-6 wks and 6 months. DMSA indicated if dilatation persists. High risk pathway – Renal USS 4-14 days + DMSA at 3 months. |
|
Referral/ Discharge |
Low risk pathway – Discharge after 6 months if USS findings resolve. Referral to Nephrology/Urology if findings persist High risk pathway - Referral to Nephrology/Urology |
|
Prophylaxis |
High Risk pathway only |
|
Genetics |
Referral/ testing not indicated. |
|
Other Monitoring |
No routine monitoring for low risk pathway High risk pathway – may require additional monitoring for severe dilatation |
Dilatation of the renal pelvis detected on antenatal ultrasound examination has an incidence of between 0.5 and 1% and may be associated with significant renal disease in a small number of babies1. However in the majority of babies the condition is benign and may be classed as isolated renal pelvis dilatation. The challenge, within the population of babies scanned antenatally, is to identify the small number of babies who have significant renal disease requiring later surgery or long term follow-up of renal function without subjecting large numbers of ‘normal’ neonates to unnecessary investigations and antibiotics. Some conditions may require urgent management such as obstruction to urinary flow- posterior urethral valves and pelviureteric junction obstruction (PUJ). Vesicoureteric reflux (VUR) is more common and the benefits of early detection are not clear2. Although there are varied approaches between centres as to the optimum method of antenatal classification, investigation and follow-up, recently there has been a gradual shift upwards in the cut-off value for investigation of renal pelvis dilatation. The following approach reflects the outcomes of recent systematic literature review.
Antenatal Screening for pelvis dilatation
An assessment of the renal system should be mandatory in all pregnancies undergoing a detailed scan and include-
- An assessment of the presence, size and morphology of each kidney
- An exact measurement of the AP pelvic diameter in mm
- An assessment of the presence or absence of calyectasis
- An indication of unilateral or bilateral disease
- An assessment of the bladder wall and distal ureters
- Documentation of any parenchymal abnormality
(e.g. renal cortical thinning, lack of corticomedullary differentiation)
Ultrasound scans should be performed by an experienced sonographer.
Details of renal tract, including the dimensions of each renal pelvis & the presence of any of the features listed above, should be clearly documented in the mother’s notes and in the paediatric communication section. The date of first detection and change over time should be recorded and made available to the paediatric team.
Parents should receive clear advice as to the nature of the problem seen and the course of events following birth. Parent information sheets are available with this guideline (Appendix).
The paediatric team should be informed antenatally if the scan indicates that the baby will fall into the high risk pathway, and there is a need for early investigation (right hand side of the flow chart below). Antenatal counselling of the family by an experienced paediatrician is desirable in these cases.
Cut-off AP measurement for investigation and prophylaxis
Traditionally, a renal pelvic diameter (RPD) cut-off of 5-7mm has been accepted in the second trimester with wide ranging reports of 5mm to 20mm3-7. The most recent review and meta-analysis of the literature, while recognising the limits of mainly observational studies, concludes that isolated AP dilatation less than 12mm has not been associated with significant morbidity and resolved in all cases studied8. Other recent large UK studies have reported little requirement for later intervention other than surveillance for dilatation below 12mm9. At levels above 12 mm the literature is too varied to allow for meaningful analysis however it is clear that an RPD above 15mm confers a higher risk for significant obstruction to urinary flow 1,11,12. However, local audit has shown that infants with normal or low risk RPD but who, in addition, have one or more of the listed risk factors have a higher incidence of problems and must be treated as per the high risk pathway13.
Antibiotic prophylaxis
The benefit of antibiotic prophylaxis to prevent breakthrough infections in this population is unclear. In the mild isolated group of babies, with RPD10-15mm, there is little evidence that picking up mild vesicoureteric reflux at this stage makes a difference to long term outcome and therefore Trimethoprim prophylaxis is not indicated10. In cases where the baby is at a high risk of urinary obstruction (RPD>15mm) or where there is already evidence of renal damage, antibiotic prophylaxis should be commenced at birth pending results of early imaging. With all babies, appropriate advice must be given to parents on the importance of urine testing if the baby is unwell and prompt treatment of infection if detected. (see appendix)
Timing of follow-up
- Babies in the high risk group with an RPD >15mm or with other risk factors -
calyectasis, parenchymal abnormality, ureterocoele, bladder wall thickening, oligohydramnios, bilateral disease etc require review by a neonatal consultant and rapid USS in the first 2 weeks of life. (N.B. an USS before 72 hours can be misleading and should be avoided if possible. However a small group of children who may require urgent intervention will require the scan in this time period – e.g. Posterior urethral valves). Following confirmation of significant RPD>15mm on USS or other risk factors, as detailed, a DMSA request should be followed by verbal and written referral to nephrology consultant.
- With milder dilatation serial US scans have been shown to be as effective as an MCUG to exclude significant VUR 4,5.
- Babies in the lower risk category should be referred for renal USS at 6-8 weeks age and 6 months age.
- Review of 1st USS will decide the need for further investigation of renal function (DMSA)
Exit from screening
A clear endpoint to follow up is desirable with most patients being categorised as ‘normal’ or ‘requiring further investigation and a renal/urological review’. A third category of baby may be followed up in a general neonatal/paediatric clinic if the DMSA is normal and the dilatation remains between 12-15mm with further investigation and treatment at the discretion of the consultant involved. Please note that in older children a pre- and post-micturition USS should be requested to exclude significant dilatation.
Parent information
Parents should have access to straight forward explanations of the condition at every stage. Information leaflets emphasize the reason for investigation, the reason for prophylactic antibiotics when prescribed and the consideration of urinary tract infection as a cause of illness. (appendix)
Flow Chart For Management of Antenatal Renal Pelvis dilatation
Patients in the highest risk category should already have been discussed with urology and may have an individualised care plan recorded in the notes.
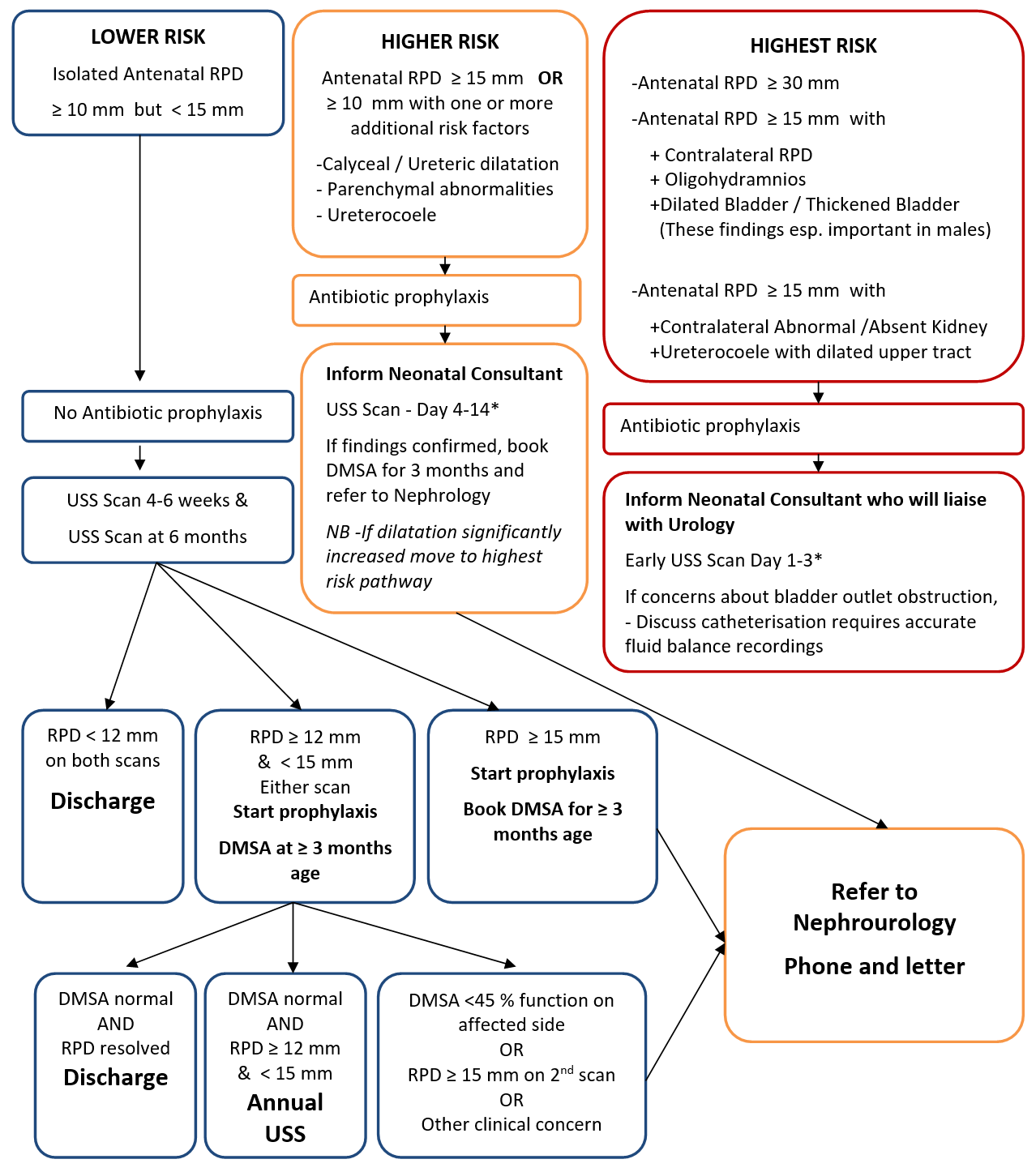
* An USS too early in the first week of life may underestimate the degree of dilatation but may be required for Highest Risk patients if there is suspicion of significant obstruction e.g Urethral valves.
Pelvi-Ureteric Junction (PUJ) Obstruction
|
Antenatal Counselling |
Consider neonatology counselling for low risk pathway (see RPD flowchart) Neonatology counselling for high risk pathway (see RPD flowchart) with Nephrology/Urology input. |
|
Attendance at Delivery/ Admission |
Not required routinely |
|
Imaging |
Renal USS – Urgency indicated as per RPD guideline |
|
Referral |
Refer to nephrourology per indications in RPD guideline. Ongoing neonatal/ medical follow up as per RPD guideline where nephrology referral not yet indicated. |
|
Prophylaxis |
|
|
Genetics |
Referral not indicated |
|
Other Monitoring |
None required |
This is the commonest cause of upper urinary tract obstruction. Up to 40% may be bilateral. The majority are primary, due to a dysfunctional segment at the PUJ, which is narrower than normal surrounding ureter. Secondary PUJO occurs due to aberrant lower pole vessels crossing the pelvis which may cause obstruction. Incidence is quoted around 1 in 1000-1500, male:female ratio is 2-3:1. The majority of cases are detected antenatally, otherwise they present with UTI, loin/abdominal pain, haematuria or a palpable flank mass. The renal pelvis and calyces are dilated to a varying degree with no ureteric dilatation. The renal pelvis may be disproportionately dilated compared to the calyces. If the calyces are not dilated it is unlikely to be obstructed. PUJO may be intermittent, depending on urine flow rate. There can be an association with other anomalies, including ectopic kidneys and duplex kidneys (lower pole).
Initial management is the same as that for RPD. Approximately 25% will require surgical intervention with pyeloplasty, which is more likely if RPD >30mm. Indications for surgery include the following:
- Reduced function <40%
- Decreasing function >10%
- RPD >40mm
- Increasing dilatation
- Symptoms such as UTI.
Often surgery will be deferred but in certain circumstances decompression may be required in the neonatal period to preserve renal function with definitive surgery offered once function confirmed. If there are any atypical features they should be discussed urgently. Other triggers for urgent discussion would include: Solitary kidney with PUJ, bilateral disease, significant dilation >30mm, significant progression on sequential scans.
Vesico-Ureteric Junction (VUJ) Obstruction
|
Antenatal Counselling |
Consider neonatology counselling for low risk pathway (see flowchart) Neonatology counselling for high risk pathway See flowchart with Nephrology/Urology input. |
|
Attendance at Delivery/ Admission |
Not required routinely |
|
Imaging |
Renal USS – Urgency indicated as per RPD guideline See table and flowchart. |
|
Referral |
Refer to nephrourology per indications in RPD guideline. Ongoing neonatal/ medical follow up as per RPD guideline where nephrology referral not yet indicated. |
|
Prophylaxis |
Indicated as per RPD guideline. See See table and flowchart. |
|
Genetics |
Referral not indicated |
|
Other Monitoring |
None required |
Obstruction at the VUJ may cause ureteric dilatation and hydronephrosis, often the degree of renal pelvis and calyceal dilatation is less than with PUJ obstruction. VUJ obstruction can also occur in association with a megaureter, either obstructed, or obstructed and refluxing. Distinction cannot be made on USS alone, although the distal ureter dilatation is often more prominent in VUJ obstruction, with tapering at the VUJ. Many will improve spontaneously, and only approximately 20% require surgical intervention such as a JJ stent or ureteric reimplantation.
Ureterocoele
|
Antenatal Counselling |
Refer to the neonatal team for discussion with family regards plan for postnatal imaging. Consult with urology where there is associated RPD ≥15mm or bilateral findings. |
|
Attendance at Delivery/ Admission |
Paediatric team not routinely required at delivery. BILATERAL RPD - Admission to NNU. Admission can be deferred for 2-3 hours if clinical condition allows. UNILATERAL RPD (CONTRALATERAL RENAL PELVIS <10mm) – Admission not required. |
|
Imaging |
BILATERAL RPD Request postnatal USS at 24-72 hours of age following urgent referral to urology. UNILATERAL RPD (CONTRALATERAL RENAL PELVIS <10mm) Postnatal USS day 4-7 |
|
Referral |
Refer to urology non-urgently when finding of uretercoele confirmed. Immediate referral required in cases with RPD ≥15mm (Unilateral or bilateral or any suggestion of changes in the contralateral kidney). |
|
Prophylaxis |
Commence prophylaxis after delivery |
|
Genetics |
Referral not indicated |
|
Other Monitoring |
BILATERAL RPD ONLY – Monitor urine output, BP, daily U&E. |
A ureterocoele is a cystic dilatation of the intravesical portion of the ureter that occurs in the bladder wall. They are more commonly associated with a duplex system with a tendency to occur in the ureter draining the upper pole of the kidney. The most common mode of presentation used to be febrile UTI but improved antenatal scanning has increased the number of asymptomatic neonates detected with this condition. In some cases a duplex system raises the possibility of a ureterocoele that is subsequently detected on postnatal imaging. As a result a more conservative approach has been adopted when treating this condition if detected antenatally.
The goal of ureterocoele management includes control of infection, preservation of renal function and protection of normal ipsilateral and contralateral units. In one study of cases detected antenatally rates of associated VUR were up to 51% with 9% of all cases grade III-V. A non-functioning upper pole on DMSA was detected in up to 54% of cases (duplex systems) but this included infants detected ‘symptomatically’ as well as those through antenatal screening. Options for definitive management of ureterocoele depend on the presenting problem but include endoscopic puncture, excision and partial or complete reconstruction of the lower urinary tract and also heminephrectomy of the non-functioning pole.
Cases where there is associated renal pelvis dilatation (RPD) ≥15mm require more urgent assessment. These infants are most likely to have a postnatal management plan in place prior to delivery that should be adhered to. Any changes noted on the contralateral kidney should raise concern that the ureterocele is causing mechanical or functional obstruction to the bladder – this may progress rapidly post natally as the bladder becomes more active and should trigger early discussion and investigation.
Otherwise, infants should commence antibiotic prophylaxis at birth.
Urgent postnatal ultrasound should be requested on day 4-7.
If the postnatal scan is normal, antibiotics can be stopped and no further follow up is required.
When the diagnosis is confirmed infants should have an MCUG and DMSA. Refer to urology to ascertain appropriate timing for MCUG & DMSA and consideration of operative management.
Megaureter
Unilateral Megaureter
|
Antenatal Counselling |
Consider referring family to Neonatal team for discussion regards postnatal management. |
|
Attendance at Delivery/ Admission |
Not required routinely. |
|
Imaging |
Renal USS – urgent in 4-7 days. MCUG / MAG3 may be required – discuss with urology once USS results known |
|
Referral |
Non-urgent referral to urology with ultrasound findings. |
|
Prophylaxis |
Commence prophylactic trimethoprim before discharge. |
|
Genetics |
Referral not indicated |
|
Other Monitoring |
None required |
Bilateral Megaureter OR Unilateral Megaureter with absent or dysplastic contralateral kidney.
|
Antenatal Counselling |
Refer family to Neonatal team for discussion regards postnatal management. NB – In boys this appearance may suggest posterior urethral valves |
|
Attendance at Delivery/ Admission |
Attendance at delivery not required routinely. Admission to NNU will be required for accurate assessment of renal function and urine output. Note male infants will require immediate urinary catheterisation prior to urology assessment. |
|
Imaging |
Urgent renal USS – 24-48 hours. |
|
Referral |
Urgent referral to nephrology & urology within 24-48 hours. |
|
Prophylaxis |
Commence antibiotic prophylaxis after delivery. |
|
Genetics |
Referral not indicated |
|
Other Monitoring |
Daily assessment of BP, U&E’s, urine output and fluid balance pending USS and discuss with urology. |
This is an abnormally wide ureter; classed as obstructed, refluxing, obstructed and refluxing or non-refluxing/non-obstructed. Secondary megaureter is due to distal obstruction i.e. urethral obstruction, bladder outlet obstruction etc. Primary megaureter occurs with cases of obstructed and non-obstructed megaureter, where secondary causes have been excluded.
- Non obstructed: The majority of cases are non obstructed with no evidence of VUR. The exact aetiology is unclear.
- Obstructed: Functional obstruction occurs due to an aperistaltic section of the distal ureter causing an inability to transport urine.
There is an increased incidence in boys and megaureter occurs more commonly in the left ureter. 25% of cases are bilateral. If unilateral approximately 10-15% will have an absent or dysplastic contralateral kidney. Most cases are diagnosed antenatally.
Management will vary similarly to the RPD guideline (page 3) where cases with bilateral involvement require more urgent assessment.
NB Secondary megaureters may require urgent treatment of the primary cause. In male infants these findings may represent a presentation of posterior urethral valves which require urgent intervention. The antenatal findings cannot differentiate this group so in male infants urgent discussion is mandatory.
Posterior Urethral Valves
|
Antenatal Counselling |
If severe (>15mm) bilateral RPD or oligohydramnios, referral to Neonatal and Urology teams is necessary and a postnatal management plan must be documented. |
|
Attendance at Delivery/ Admission |
Attendance not required routinely unless there is severe oligohydramnios. Followed in all cases by admission to NNU – this may be deferred for 2-3 hours if the infant’s clinical condition allows. On admission all should have IV access established; baseline bloods (accepting that this will reflect maternal renal function – but important as a first point in the trend) and urinary catheterisation. Close monitoring for electrolyte changes associated with post obstructive diuresis. |
|
Imaging |
Urgent renal USS within 24-48 hours of delivery unless otherwise specified by a management plan agreed up on antenatally. Early MCUG will be required to confirm the diagnosis – discuss with urology. |
|
Referral |
Urgent referral to Urology and Nephrology if suspicion of this diagnosis or if identified at postnatal USS. May already have a pathway agreed up on in antenatal period |
|
Prophylaxis |
Commence antibiotic prophylaxis following USS confirmation or sooner if pre-agreed. |
|
Genetics |
Referral not indicated |
|
Other Monitoring |
Daily U&E’s, blood pressure, measurement of urine output and fluid balance (majority will need fluid and electrolyte replacement in the early phase). If the condition is suspected (or confirmed) establishment of bladder drainage is mandatory while other neonatal care continues and further investigations are arranged. Post catheterisation one must anticipate a post obstructive diuresis and fluid shifts which require careful fluid balance management |
Posterior urethral valves are a congenital malformation of the posterior urethra, only seen in male infants. Valves affect the developing urinary tract from an early stage, and lead to bladder outlet obstruction which can cause VUR, hydronephrosis, and calyceal rupture. Valves can lead to pulmonary hypoplasia if oligohydramnios has developed secondary to urinary obstruction. The degree of renal impairment depends on the degree of renal dysplasia, either integral to the condition, or secondary to obstruction. A significant proportion of these infants will progress to end stage renal failure before adulthood despite aggressive and appropriate therapy. Posterior urethral valves is a chronic condition requiring life long monitoring and treatments. Antenatal treatments have been proposed for valves boys but at present the data does not support improvement in renal outcomes sufficient to justify routine intervention outside trials.
Posterior urethral valves are exclusive to male infants. Rarely anterior urethral valves affect females. Estimated incidence 1 in 5000 –12000. 40-60% are seen on antenatal USS with oligohydramnios, bilateral hydronephrosis and hydroureters, bladder wall thickening and posterior urethral dilatation. Occasionally PUV’s can be missed without a third trimester scan. 10% of boys with posterior urethral valves have unilateral hydronephrosis, as such the diagnosis must be considered even with unilateral changes.
Oligohydramnios can cause postural defects such as talipes, dislocated hip, receding jaw ie Potter like syndrome. Severe oligohydramnios and resulting pulmonary hypoplasia can be fatal in the neonatal period.
Renal function at birth can vary from normal to severely impaired. Tubular damage leads to sodium and bicarbonate wasting, with secondary hyperkalaemia. Following surgical treatment of valves and relief of obstruction, creatinine may normalise, or remain significantly elevated, dependent on the degree of renal dysplasia.
Cases with significant antenatal bilateral hydronephrosis and/or oligohydramnios should be discussed antenatally with urology and a postnatal plan documented.
If there is bilateral hydronephrosis and suspicion of PUV’s, arrange an urgent USS within the first 48 hours. Discuss with the neonatal consultant and urology. Urinary catherisation should be performed in all cases to relieve obstruction pending further imaging and investigation.
MCUG is the gold standard imaging for diagnosis. It will show posterior urethral dilatation and thickened bladder wall with an abnormal trabeculated contour, unilateral or bilateral VUR may be present. In cases where MCU is non diagnostic or were the index of suspicion is high it may still be appropriate to consider cystoscopy. It is possible for cases of PUV’s not to be detected on MCUG especially where there is high grade reflux.
While awaiting surgery following catheterisation there needs to be careful attention to fluid and electrolyte balance as post obstructive diuresis occurs and these babies will often need additional sodium and bicarbonate. Joint management with Nephrology is essential to optimise the renal recovery.
Treatment is surgical. The valves are ablated cystoscopically. If valve ablation is not possible urinary diversion with vesicostomy or bilateral ureterostomies may be needed with later valve surgery and subsequent recontruction.
Long term follow up will be shared by the Nephrology and Urology teams. Ongoing USS monitoring for renal tract dilatation will be required. Repeat cystoscope or MCUG at approximately three months is needed to ensure there are no residual valve leaflets.
DMSA at approximately three months after relief of obstruction and resolution of infection is needed to assess the degree of renal dysplasia, along with monitoring of renal function. However, outcomes should remain guarded and life long monitoring will be required. Normal renal function at 2 years of life is a good prognostic sign but even in this group CKD in later life is expected.
Familial Vesicoureteric Reflux (VUR) – Screening siblings of affected children.
Screening asymptomatic siblings of affected children.
|
Antenatal Counselling |
Not routinely required. |
|
Attendance at Delivery/ Admission |
Not routinely required. NNU admission not required. |
|
Imaging |
Renal USS 4-6 weeks – Follow RPD guideline with findings. |
|
Referral/ Discharge |
Pending Postnatal USS findings. Indicated as per RPD guideline. See See table and flowchart. |
|
Prophylaxis |
Only if renal pelvis dilatation detected antenatally. Indicated as per RPD guideline. See table and flowchart. |
|
Genetics |
Referral/ testing not indicated. |
|
Other Monitoring |
None required. |
Vesicoureteric reflux in children has a prevalence of 1-2%. In the majority of cases the degree of reflux is mild and such cases commonly self resolve later in childhood. However there is a smaller group with more severe VUR who are at increased risk of complications such as pyelonephritis and renal scarring.
The prevalence of VUR is known to be increased within families. Recent studies show an increase in prevalence to 27.4% amongst children with an affected sibling or 35.7% amongst children with an affected parent. The prevalence is higher if screening is performed at < 2y of age as the condition naturally improves with age. High grade reflux (Grade III – V) is identified in a minority (9.8% of those screened) confirming that severe reflux, within families, is less common. Renal cortical abnormalities were identified in 14.5% of screened individuals without prior UTI. Overall (symptomatic and asymptomatic) the prevalence amongst family members screened was 22.5%.
Screening 1st degree relatives of those with VUR may have the potential to facilitate earlier identification and if necessary earlier intervention for individuals with VUR. However, due to a lack of prospective randomised studies it is unclear whether such screening will be effective in preventing UTI’s and renal scarring in the long term.
For the purposes of this guideline the following recommendations are made
- Screening should be offered where there is a family history, in a first degree relative, of high grade reflux (Grade III-V) or reflux associated with urinary tract infection or renal scarring. Screening by USS should be offered as a default if the grade of reflux in the relative is unknown.
- Screening should be with Renal ultrasonography at 4-6 weeks
- Additional investigations are only warranted if there are abnormalities on the renal ultrasound. Refer to relevant sections of this guideline
- If the infant is the index case then screening may be recommended for older siblings who are not yet toilet trained. (there is no additional benefit to screening older children who are toilet trained)
Duplication of the Ureters (Duplex Collecting System/ Duplex Kidney(s))
|
Antenatal Counselling |
Consider referral to neonatal team for discussion with family regards plan for postnatal imaging. |
|
Attendance at Delivery/ Admission |
Not routinely required. |
|
Imaging |
Unilateral involvement – Renal USS 4-6 weeks Bilateral involvement – Renal USS 4-7 days Associated RPD – follow RPD guideline. See table and flowchart. |
|
Referral/ Discharge |
Referral to nephrology team if: 1. Evidence of RPD on postnatal USS – follow RPD guideline. See table and flowchart. Ongoing Neonatal/ medical follow up: 1. If renal USS does NOT demonstrate RPD or any other abnormality other than a unilateral or bilateral duplex system - DISCHARGE |
|
Prophylaxis |
Not required unless renal tract dilatation present on antenatal or postnatal USS. See RPD Guideline table and flowchart. |
|
Genetics |
Referral not indicated. |
|
Other Monitoring |
None required. |
A duplex or duplicated system is defined as a kidney with two pelvicalyceal systems with or without duplication of the ureter. In incomplete duplication, both ureters join before entering the bladder with a bifid pelvis in the mildest form. In cases of complete duplication, two ureters enter the bladder separately. Incomplete duplication is more common than complete duplication in a ratio of 3:1, with a reported incidence of all forms of 1:125 individuals. Rarely, there may be bilateral duplex systems.
Less often, there are associated renal abnormalities include vesicoureteric reflux, PUJ obstruction (much less likely in complete duplication) and in complete duplication alone, ectopic ureterocoele or ectopic ureteral insertion.
Infants with an antenatal diagnosis should receive a postnatal ultrasound urgently for bilateral duplex kidneys (4-7 days) and routinely for unilateral duplex systems (4-6 weeks). Cases should be considered more urgently where there is renal pelvis dilatation found antenatally. Prophylactic antibiotics are indicated as per the RPD guideline.
If, following postnatal ultrasound, there are other findings, such as pelvicalyceal or ureteric dilatation follow the RPD guideline regards further imaging and referral. See table and flowchart.
Contents:
- Single Cyst/ Simple Renal Cyst
- Multicystic Dysplastic Kidney (MCDK)
- Foetal hyperechogenic kidneys (isolated finding)
- Autosomal Recessive Polycystic Kidney Disease (ARPKD)
- Family History of ARPKD
- Autosomal Dominant Polycystic Kidney Disease – Family History of,
Single Cyst/ Simple Renal Cyst
|
Antenatal Counselling |
Not required unless there is a family history of cystic renal disease. |
|
Attendance at Delivery/ Admission |
Not required routinely |
|
Imaging |
Non-urgent renal USS at 4-6 weeks. |
|
Referral |
Refer to nephrology if: 1. Findings persist at 4-6 weeks with associated RPD OR multiple cysts. Follow RPD guideline if relevant - table (Page 3) and flowchart (page 5). 2. If finding of a single cyst persists beyond 1 year of life. Ongoing neonatal/ medical follow up 1. Normal scan – DISCHARGE 2. If findings persist at 4-6 weeks repeat USS at 1 year of age |
|
Prophylaxis |
Not required unless renal tract dilatation present on antenatal or postnatal USS. See RPD Guideline, table (Page 3) and flowchart (page 5). |
|
Genetics |
Referral not indicated |
|
Other Monitoring |
None required |
Renal cysts may be multiple or solitary. Multiple cysts may represent ARPKD or MCDK (mentioned elsewhere in this guideline). In cases of solitary renal cysts the majority are simple (i.e. non loculated, non-communicating with normal gross appearance of the kidney and no obstruction to the pelvis or ureter). They are often benign. In one series, of 28 cases detected antenatally, 25 (89%) resolved during pregnancy. 1 was later redefined as MCDK as there were multiple cysts evident on further follow up. The remaining 2 persisted to 3 & 6 years.
If persistent it is still likely that most simple renal cysts will spontaneously resolve after delivery.
- Rule out a family history of cystic renal disease.
- Non urgent post natal ultrasound (4-6 weeks) is required.
- If normal then discharge from follow up
- If still present repeat at 1 year – if persists discuss with renal team.
- If family history positive or multiple cyst or associated renal abnormalities discuss with the renal team.
Multicystic Dysplastic Kidney (MCDK)
1. Normal contralateral kidney/ RPD of contralateral kidney <15mm
|
Antenatal Counselling |
Offer counselling with Neonatal and Nephrology team to discuss requirement for postnatal imaging |
|
Attendance at Delivery/ Admission |
Not routinely required. |
|
Imaging |
Normal contralateral kidney - Ultrasound 4-6 weeks & DMSA at 3 months RPD of contralateral kidney - see table (Page 3) and flowchart (page 5). |
|
Referral/ Discharge |
Refer to nephrology team if:
Ongoing Neonatal/ medical follow up if:
Neonatal/ medical team to arrange renal USS at 12 months:
|
|
Prophylaxis |
Not required unless renal tract dilatation present on antenatal or postnatal USS, see table and flowchart in section above. |
|
Genetics |
None required. |
|
Other Monitoring |
None required |
2. Abnormal contralateral kidney (including RPD ≥ 15mm and/or cystic changes) OR oligohydramnios
|
Antenatal Counselling |
Offer counselling with Neonatal team to discuss requirement for admission and urgent postnatal imaging. |
|
Attendance at Delivery/ Admission |
Neonatal team to attend delivery. Admission to NNU within 2-3 hours if clinical condition at delivery allows. |
|
Imaging |
Urgent postnatal USS 48-72 hours |
|
Referral/ Discharge |
Urgent inpatient referral to nephrology team |
|
Prophylaxis |
Not required unless renal tract dilatation present on antenatal or postnatal USS (See RPD Guideline) |
|
Genetics |
Offer Clinical Genetics referral to the family. There are known single gene causes of bilateral renal cystic changes for which testing is available. This may prompt further follow up as other organ systems can be affected. |
|
Other Monitoring |
Daily blood pressure, U&E, urine output and fluid balance pending USS imaging and nephrology advice. |
MCDK is a congenital developmental anomaly with an incidence of around 1:4,300 live births. A multicystic dysplastic kidney has no functioning renal tissue and bilateral involvement is incompatible with life.
One series demonstrated that over 40% of MCDK’s involute spontaneously with a further 30% involuting partially and the remainder found to be unchanged during follow up. The same study showed the contralateral kidney may show compensatory hypertrophy (77%) which is a normal finding. Alternatively, the kidney may be of normal size (6%), this may suggest dysplasia and requires long term follow up. Occasionally the contralateral kidney may show mild hydronephrosis (10%), or obvious dyplasia (2%).
Contralateral renal anomalies, most commonly VUR (mostly mild and self resolving), but also vesicoureteric/ pelviureteric junction obstruction and ureterocoele may be found in up to 16% of cases. Despite this, occurrence of UTI was low. As the MCDK is non functional, any abnormality to the effectively solitary kidney needs more aggressive assessment and management than may be indicated in normal unilateral situations.
In uncomplicated cases, long term radiological follow up of patients with MCDK is necessary to track involution of the affected kidney where this has not occurred antenatally AND to ensure compensatory hypertrophy of the unaffected kidney.
(Historically there has been a suggestion of risk of neoplasia in the affected kidney - Wilm’s tumour. A large meta analysis found no such association exists and that ultrasound screening would be needed at least every 3 months to detect neoplasia. Rates of hypertension in these individuals were found to be similar or less than the background prevalence that exists in the general paediatric population, 5.4/1000 cases). The main indications for surgery are failure of involution and family preference for removal reducing need for follow up or atypical features suggesting incorrect diagnosis and reduced likelihood of involution.
1. Normal contralateral kidney/ RPD of contralateral kidney <15mm on antenatal scans.
Normal contralateral kidney - Routine renal ultrasound at 4-6 weeks & DMSA at 3 months to identify if there is any functioning renal tissue – MCDK is totally non-functioning.
RPD of contralateral kidney <15mm – Follow guidance in table and flowchart in section above.
Referral to the nephrology team should occur when:
- There is any abnormality of the contralateral kidney on ultrasound (i.e. dilatation or cysts) or DMSA – URGENT REFERRAL BY PHONE
- There is a remnant of MCDK present on postnatal ultrasound (i.e. involution has not occurred) – Written referral.
MCUG is NOT routinely required. This has been advocated in the past. Only postnatal ultrasound findings indicate if MCUG is required or not in these cases.
Ongoing Neonatal/ Medical follow up should occur with the following findings:
- Renal USS – Involution of affected kidney AND no abnormality of contralateral kidney.
- DMSA – Normal function of contralateral kidney
Arrange renal USS at 12 months. Refer to nephrology non-urgently where compensatory hypertrophy is NOT demonstrated. Where there is compensatory hypertrophy, discharge from neonatal/ medical follow up. Current evidence would suggest that there is no risk of neoplasia in the affected kidney.
2. Abnormal contralateral kidney or oligohydramnios
Bilateral MCDK is incompatible with life as there is associated oligohydramnios and severe pulmonary hypoplasia. The parents should be counselled to expect that their baby is unlikely to survive for very long. Plans should be made for comfort care for the infant following delivery.
If a single kidney is affected by MCDK but there are contralateral changes – eg parenchymal cysts or pelvicalyceal dilatation ≥15mm, OR if there are concerns about renal function due to the presence of oligohydramnios then the infant will require urgent investigation and close monitoring. This should include: Daily blood pressure; U&E; urine output and fluid balance. An early (48 – 72) USS should be arranged and the case should be discussed with the Nephrologists. Genetic referral would be warranted as these changes may be associated with HNF1β gene mutations.
Foetal hyperechogenic kidneys (isolated finding)
|
Antenatal Counselling |
ONLY if there are associated findings i.e. size >95th centile, cystic changes reduced liquor or family history of renal disease. There is a greater likelihood of renal disease being present in these cases. |
|
Attendance at Delivery/ Admission |
Not required |
|
Imaging |
Postnatal USS at 4-6 weeks |
|
Referral/ Discharge |
Use recommendations elsewhere in this guideline based on postnatal findings. For ongoing/ inconclusive finding of ‘bright kidneys’ alone, refer non-urgently to nephrology team. |
|
Prophylaxis |
Not required unless renal tract dilatation present on antenatal or postnatal USS see table and flowchart in section above. |
|
Genetics |
Discuss referral to the local Clinical Genetics Service with the family ONLY if there are one OR more of the following features present on postnatal USS
|
|
Other Monitoring |
In cases associated with a large placenta consider monitoring urine output U&E’s & urine dipstick for protein content for 24-72 hours after birth for cases of congenital nephrotic syndrome. |
The management of foetal hyperechogenic kidneys remains controversial. Kidney hyperechogenicity is defined as kidneys that appear more echogenic than the liver or spleen after 17 weeks gestation.
Multicystic dysplastic kidneys appear hyperechoic but have associated structural anomalies.
In ARPKD there are usually (but not always) cystic changes, the kidneys may appear large (>95th centile) and there may be reduced liquor volume.
Mutations in the HNF1-β gene may also produce echogenic kidneys with associated findings therefore it may be necessary to refer the family for genetic surveillance.
ADPKD may present with bright kidneys on antenatal scan and in some cases cysts may be seen, although in most these will not be apparent until later in life. The kidneys are not enlarged.
A retrospective study looked at 93 foetuses presenting with hyperechogenic kidneys (in the absence of other renal tract anomalies). This study included, only those, with a diagnosis of nephropathy confirmed later. In this series 28/93 had ADPKD, 31/93 ARPKD and 34/93 associated syndromes (Bardet-Biedl, Meckel-Gruber, Trisomy 13 and others). On detailed ultrasound examination there were cystic changes evident in 30/93.
In a much smaller series of 7 foetuses with hyperechogenic kidneys and normal liquor volume, only one infant was found to have normal kidneys. Older series in the last 30 years have quoted figures likewise.
Existing data suggests that, in isolation, the finding of hyperechogenic kidneys still bares significance but is not pathognomic of renal disease. Antenatally, appropriate family history is helpful and occasionally genetic survey and ultrasound of the parents may be deemed appropriate to determine significance.
In any case postnatal ultrasound at 4-6 weeks is necessary to rule out any abnormality. Antibiotic prophylaxis is not required unless renal tract dilatation is diagnosed antenatally, in which case follow the RPD guideline.
Autosomal Recessive Polycystic Kidney Disease (ARPKD)
|
Antenatal Counselling |
Refer family to Neonatal and Nephrology teams for antenatal counselling. |
|
Attendance at Delivery/ Admission |
Neonatal team to be present at delivery (primarily to assess requirement for respiratory support). Followed by admission to NICU which may be deferred for 2-3 hours if the infant’s clinical condition allows. |
|
Imaging |
Urgent renal USS (24-48 hours) for baseline assessment of kidney size (AP diameter will be more accurately assessed on repeat scans). Further imaging pending acute clinical course and discussion with nephrology team. |
|
Referral |
Urgent referral to nephrology same/ next day. |
|
Prophylaxis |
For those with a milder phenotype who are deemed likely to survive to discharge from NNU, prophylaxis should be offered. |
|
Genetics |
Discuss referral to Clinical Genetics Service for genetic testing with family. Some polycystic kidneys in neonatal period have an underlying syndromal cause and genetic testing will be based on phenotype. |
|
Other Monitoring |
Daily assessment of U&E’s, BP, urine output and fluid balance. |
ARPKD has a reported incidence of between 1:10,000 & 1:55,000 births. Both kidneys are affected by cysts which develop from dilated collecting ducts. There is often hepatic involvement by the same mechanism and the severity varies inversely with that of the renal disease. There is variable genetic expression producing a wide spectrum of illness with varying degrees of renal dysfunction. In the antenatal period, oligohydramnios and compression of the lungs by the enlarged kidneys contribute to varying severities of pulmonary hypoplasia. Dependent on severity, infants can succumb to respiratory rather than renal complications. However, survival rates overall at 1 year of age are reported around 87%.
In a retrospective case series of 31 patients (10 diagnosed prenatally) the highest mortality was amongst those diagnosed prenatally or in infancy, typically due to respiratory failure. Renal impairment was of greater severity with less likelihood of long term improvement. Other complications include the following:
- Chronic Renal Insufficiency. 7/13 (54%) neonates were found to have mild to moderate renal insufficiency, the remainder having normal renal function. Renal impairment was strongly associated with pulmonary insufficiency. There was no requirement for renal replacement therapy (i.e. peritoneal dialysis) in the first year of life.
- Hypertension. 11/13 (85%) neonates developed hypertension (age at diagnosis 4 days – 36 months).
- Electrolyte abnormalities. Hyponatraemia was documented in 3 infants diagnosed in the immediate postnatal period. This was self resolving after 1 month.
- Others. Rates of UTI and VUR were low with 6 & 1 cases respectively in the entire series of 31 infants and children.
It is most likely that infants diagnosed with ARPKD in the antenatal period will have been discussed with the relevant subspecialties in advance. Such infants may require respiratory support at birth and the paediatric team would be present at delivery for immediate assessment.
Thereafter renal management includes: Daily monitoring of U&Es and creatinine, Daily weight, Monitoring urine output and fluid balance, Monitoring of blood pressure (non invasive or otherwise)
Immediate discussion with the nephrology team.
Baseline assessment of kidney size by renal ultrasound in the first few days of life.
Simultaneous assessment of liver and gall bladder by abdominal ultrasound.
Family History of ARPKD
|
Antenatal Counselling |
Family may wish to discuss requirement for postnatal USS with neonatal team in cases where this will be indicated. |
|
Attendance at Delivery/ Admission |
Not required. |
|
Imaging |
Urgent postnatal USS day 4-14. |
|
Referral |
Urgent referral to nephrology team if abnormal kidneys identified on USS. Further investigation by nephrology team required to confirm findings. |
|
Prophylaxis |
Not required unless renal tract dilatation present on antenatal or postnatal USS, see table (Page 3) and flowchart (page 5). |
|
Genetics |
Consider referral to local clinical genetics if not already done on account of affected relative OR refer if confirmatory findings on postnatal scan. |
|
Other Monitoring |
None required. |
Where there is an affected sibling the risk to the infant is 1:4. It is possible there may have been prenatal testing performed in the antenatal period where there is an affected sibling. Referral for non urgent postnatal renal & abdominal ultrasound should occur in the following instances:
- Where there is an affected sibling AND no apparent abnormality antenatally or on postnatal physical examination AND there has been no prenatal testing.
- Where there is an affected parent and the parents are consanguineous with no apparent abnormality antenatally or on postnatal physical examination.
Subsequent referral to the nephrology team is required where there are abnormal findings.
In cases where there is an affected parent (non-consanguinous) or other non-1st degree relative it is highly unlikely a child will be affected by this condition.
Autosomal Dominant Polycystic Kidney Disease – Family History of,
|
Antenatal Counselling |
Referral to the neonatal team for counselling may be useful in cases where there are USS changes or an unconfirmed family history. |
|
Attendance at Delivery/ Admission |
Not required routinely. |
|
Imaging |
Non-urgent USS 4-6 weeks in cases where there are antenatal USS changes or unconfirmed family history. USS NOT routinely indicated where the there are no abnormal antenatal findings and the family history is not in doubt. |
|
Referral/ Discharge |
Refer to nephrology team if: 1. Cystic or other changes on renal USS. Ongoing Neonatal/ medical follow up. 1. No ongoing follow up required unless there is a decision to perform USS 2. If decision to scan and USS normal – discharge from neonatal care. (These infants may be followed up by genetics) |
|
Prophylaxis |
Not required unless renal tract dilatation present on antenatal or postnatal USS see table and flowchart in section above. |
|
Genetics |
If not already done so on account of affected relatives, discuss referral to Clinical Genetics Service with the family as there are known gene defects (PKD1 & PKD2 genes) with testing available. On confirmation, ‘cascade’ testing may be offered to other family members. Alternatively, the family may wish to wait until the child is old enough to make their own informed decision regards testing and investigation. |
|
Other Monitoring |
None required. |
This is the commonest inherited kidney disease with an incidence of 1:500 to 1:1000 with clinical presentation found mostly in adults. The cysts originate from the epithelia of the nephron and collecting ducts and cystic changes are focal rather than diffuse as with ARPKD. It is possible for this condition to present in childhood with symptoms including, flank pain, haematuria, abdominal masses, hypertension and other, non-renal, manifestations.
Genetic testing is available but is not always conclusive, therefore ultrasound imaging is still a key component to making the diagnosis. The radiological diagnosis is age-dependent and it is difficult to exclude a diagnosis of ADPKD with USS alone until a ‘normal’ USS result is obtained at/after 30 years of age.
Unless there are any antenatal abnormalities i.e. cystic changes or bright kidneys OR the family history is unclear, there is no requirement for routine screening or follow up in the neonatal period.
Contents:
- Single kidney / Absent kidney
- Pelvic kidney/ Ectopic Kidney
- Renal Ectopia/ Cross Fused Renal Ectopia
- Horseshoe Kidney
- Bilateral Renal Agenesis
- Enlarged Kidney/ Asymmetrical Kidney Size
Single kidney / Absent kidney
|
Antenatal Counselling |
Consider referral to neonatal team for discussion with family regards plan for postnatal imaging. |
|
Attendance at Delivery/ Admission |
Not routinely required. |
|
Imaging |
Postnatal USS 4-6 weeks & DMSA at 3 months. Arrange MCUG if renal tract dilatation on USS or abnormal DMSA. |
|
Referral/ Discharge |
Refer to nephrology if:
Ongoing Neonatal/ Medical follow up.
At 12 months, renal USS shows NO compensatory hypertrophy – non-urgent referral to the nephrology team. |
|
Prophylaxis |
Only indicated if dilatation or other risk factors as per RPD guideline, see table and flowchart in section above (Obstruction/dilation of Renal Tract). |
|
Genetics |
If other congenital anomalies are present, including deafness, discuss referral to the Clinical Genetics Service with the family. Underlying causes include known single gene and chromosomal defects for which testing is available. This would allow accurate recurrence risks for future pregnancies to be determined. |
|
Other Monitoring |
None required. |
True unilateral renal agenesis (URA) has an estimated incidence of 1 in ~2000 births. It is defined as the congenital absence of one kidney as a result of failure of embryonic development. Unrecognised ectopic kidney and a multicystic dysplastic kidney that has involuted, may give similar appearances antenatally.
The most recent systematic review URA was associated with other renal anomalies in 32% of cases. VUR was the commonest abnormality (24%), other conditions included, megaureter (7%), PUJO (6%), duplex kidney (3%), ureterocoeles (1%) & PUV (1%).
On this basis a postnatal ultrasound would be recommended (4-6 weeks) to help confirm the antenatal findings and establish if associated anomalies are present. Thereafter the infant should receive a DMSA to look for ectopic renal tissue. With a solitary kidney with any abnormal antenatal findings early discussion with nephro-urology would be appropriate and earlier investigation may be indicated.
In the presence of a normal contralateral kidney on postnatal USS and evidence of true URA on DMSA, follow up at one year is recommended. Where there is no compensatory hypertrophy (see Appendix 1) of the remaining kidney at 12 months, there should be a non-urgent referral to the nephrology team.
An MCUG would be recommended on the basis of any abnormal findings in the remaining kidney on the postnatal USS or DMSA and subsequent referral to the nephrology team. Commence prophylactic antibiotics where there is dilatation of the renal pelvis, see RPD guideline.
Pelvic kidney/ Ectopic Kidney
|
Antenatal Counselling |
Not routinely indicated. |
|
Attendance at Delivery/ Admission |
Not routinely indicated. |
|
Imaging |
Routine renal USS 4-6 weeks. DMSA 3 months. |
|
Referral |
Nephrology referral if: 1. Abnormal contralateral kidney on USS 2. Renal pelvis dilatation >15mm 3. Reduced function on DMSA Ongoing Neonatal / Medical follow up if: 1. Normal contralateral kidney, normal split function on DMSA. 2. Review at 6 months & 12 months with renal USS, BP and urine dipstick. If normal – DISCHARGE. Refer to nephrology if abnormal USS/ hypertension, abnormal urinalysis. – if relevant, follow RPD guideline. see table and flowchart in section above (Obstruction/dilation of Renal Tract). |
|
Prophylaxis |
Only indicated if dilatation or other risk factors as per RPD guideline, see table and flowchart in section above (Obstruction/dilation of Renal Tract). |
|
Genetics |
Not required |
|
Other Monitoring |
Not required |
During normal embryogenesis there is a failure of ascent of the kidney. The resulting kidney is usually discoid rather than reniform in shape. An ectopic kidney can occur anywhere along the path of embryological ascent from pelvis to renal fossa, however pelvic kidneys are the most common. This occurs in 1:2200-3000 live births. 90% are unilateral with the left side more predominantly affected. Obstructed drainage, renal hypoplasia/ dysplasia or ectopic ureter can be associated. Obstruction may be present during infancy or may develop with growth as the orientation changes.
Follow up is non-urgent unless there are indications for urgent investigation and referral as per the RPD guideline. Likewise antibiotic prophylaxis is indicated only when there is antenatal evidence of RPD. DMSA is required to confirm USS diagnosis and exclude other ectopic renal tissue.
Renal Ectopia/ Cross Fused Renal Ectopia
|
Antenatal Counselling |
Consider referral to neonatal team for discussion with family regards plan for postnatal imaging |
|
Attendance at Delivery/ Admission |
Not routinely required. |
|
Imaging |
Postnatal USS at 4-6 weeks. Arrange DMSA at 3 months following confirmatory findings on USS. Arrange MCUG if renal pelvis dilatation present |
|
Referral/ Discharge |
Refer to Nephrology if: 1. Abnormal USS i.e. RPD OR abnormal function on DMSA* Ongoing Neonatal/ Medical follow up: 1. If no RPD on USS and normal function on DMSA, repeat USS at 12 months 2. If normal growth – DISCHARGE. If poor renal growth demonstrated, refer to nephrology team. *Follow RPD guideline. see table and flowchart in section above (Obstruction/dilation of Renal Tract). |
|
Prophylaxis |
Only indicated if dilatation or other risk factors as per RPD guideline, see table and flowchart in section above (Obstruction/dilation of Renal Tract). |
|
Genetics |
Referral not indicated. |
|
Other Monitoring |
None required. |
Renal ectopia is a rare condition where the kidney lies outside the renal fossa. Simple renal ectopia implies the kidney lies ipsilateral in the pelvis. In crossed renal ectopia, the kidney lies contralateral to the side where the ureter enters the bladder. In the majority of cases of crossed renal ectopia the ectopic kidney lies inferiorly with it’s upper pole fused to the lower pole of the orthotopic unit (80-90%), named cross-fused renal ectopia. Fusion is present in 85-90% of cases of crossed ectopia. The degree of fusion can be categorised but does not alter management in the postnatal period.
Incidence of this condition at post mortem is up to 1:1000 but only 1:10,000 when detected ‘symptomatically’ indicating that this condition is largely uncomplicated. In a series of 47 children diagnosed prenatally and postnatally (i.e. symptomatically or under investigation for multiple abnormalities) with ectopic kidney or crossed renal ectopia, (mean age at postnatal presentation 2-3 months) the principal associated abnormalites included VUR, PUJ obstruction and MCDK. Associated anomalies were more prevalent in simple renal ectopia with VUR more likely to affect the orthotopic unit in ‘crossed’ cases. The majority of those with VUR experienced spontaneous resolution by around 7 years of age. Relative function of the ectopic kidney on DMSA was 38%.
With the associated risks, further investigation is necessary. In the first instance there should be a postnatal ultrasound at 4-6 weeks followed by DMSA at 3 months. Where there is urinary tract dilatation on postnatal ultrasound an MCUG should be arranged with referral to the nephrology team (follow RPD guideline). Where there is an abnormality on DMSA, refer to the nephrology team.
Horseshoe Kidney
|
Antenatal Counselling |
Consider referral to neonatal team for discussion with family regards plan for postnatal imaging |
|
Attendance at Delivery/ Admission |
Not routinely required. |
|
Imaging |
Postnatal USS at 4-6 weeks. Arrange DMSA at 3 months following confirmatory findings on USS. Arrange MCUG if renal pelvis dilatation present - Follow RPD guideline. see table and flowchart in section above (Obstruction/dilation of Renal Tract). |
|
Referral/ Discharge |
Refer to Nephrology for ongoing surveillance |
|
Prophylaxis |
Only indicated if dilatation or other risk factors as per RPD guideline, see table and flowchart in section above (Obstruction/dilation of Renal Tract). |
|
Genetics |
If there is a clinical suspicion of a syndromal or chromosomal abnormality (i.e. Turners or trisomy) associated with horseshoe kidney, discuss referral to local Clinical Genetics Service with family. |
|
Other Monitoring |
None required unless there are associated anomalies. |
Horseshoe kidney is a condition that can be included on the spectrum of ‘fusion anomalies’ including cross-fused renal ectopia. It is the commonest fusion abnormality with an incidence of approximately 1:400 people. Both kidneys fuse across the midline at the lower poles. Fusion of the upper poles and much more rarely both poles (sigmoid kidney) is possible. The commonest associated renal problems include PUJ obstruction and infection. Horseshoe kidney is also associated with Turner’s syndrome, trisomies and VACTERL.
In the first instance there should be a postnatal ultrasound at 4-6 weeks followed by DMSA at 3 months. If there is any urinary tract dilatation evident order an MCUG. Refer to the nephrology team regardless of findings as ongoing follow up is required long term to ensure adequate renal growth, normal BP and no late sequelae such as PUJO.
Bilateral Renal Agenesis
This is rare and almost universally fatal, with many intrauterine deaths. Oligohydramnios occurs from around 16 weeks leading to Potter sequence. If identified antenatally, parents should be counselled appropriately, and a neonatal management plan documented. Fetuses surviving to term usually die within hours of birth due to pulmonary hypoplasia. There may be a very small number of survivors who will require management for end stage renal failure from birth.
If bilateral renal agenesis occurs as part of a multiple malformation complex there may be an underlying single gene or chromosomal cause with a recurrence risk in future pregnancies. Discussing referral to the Clinical Genetics Service with the family would be helpful.
Enlarged Kidney/ Asymmetrical Kidney Size
|
Antenatal Counselling |
Not required for enlarged kidney alone. If there is suspicion of an overgrowth syndrome then refer family to neonatal team for antenatal counselling. |
|
Attendance at Delivery/ Admission |
Not required routinely |
|
Imaging |
Non-urgent renal USS at 4-6 weeks. |
|
Referral |
Refer to nephrology if: 1. Suspicion of hemihypertrophy/ tumour Ongoing Neonatal/ medical follow up 1. If abnormality identified on renal scanning then refer to relevant section in this guideline. 2. If postnatal renal scan normal - DISCHARGE |
|
Prophylaxis |
Not required unless renal tract dilatation present on antenatal or postnatal USS - see table and flowchart in section above (Obstruction/dilation of Renal Tract). |
|
Genetics |
Referral not indicated unless diagnosis of hemihypertrophy syndrome i.e. BWS suspected postnatally. |
|
Other Monitoring |
None required |
This can be due to a duplex collecting system, obstruction, parenchymal abnormalities or rarely a tumour. Hemihypertrophy as a result of Beckwith-Wiedemann or other overgrowth syndromes is also a possible underlying cause.
Unless there are other syndromic associations, these cases require non-urgent renal USS (4-6 weeks) and findings acted upon as outlined in the relevant section of this guideline. If there RPD is detected then this should be managed as per the RPD guideline. Where renal involvement in one of the overgrowth syndromes is suspected more urgent referral is required.
Contents:
Absent Abdominal Musculature Syndrome/ Prune Belly Syndrome
|
Antenatal Counselling |
Refer family to Neonatal and Nephrology teams for antenatal counselling. |
|
Attendance at Delivery/ Admission |
The paediatric team will attend delivery for immediate assessment of respiratory status followed by admission to NNU. Admission may be deferred 2-3 hours where the infants clinical condition allows. |
|
Imaging |
Urgent renal USS within 24-48 hours. |
|
Referral |
Urgent referral to nephrology and urology teams within 24-48 hours. |
|
Prophylaxis |
Commence antibiotic prophylaxis pending further imaging. |
|
Genetics |
Some cases of prune belly syndrome have an underlying single gene defect for which testing is available. If it is possible to confirm the molecular diagnosis, accurate recurrence risks for future pregnancies can be discussed with the family. Refer to genetics service. |
|
Other Monitoring |
Daily BP, U&E, urine output and fluid balance required. Routine echocardiogram. Other investigations indicated for anomalies identified on antenatal USS or postnatal examination. |
Previously called ‘prune belly syndrome’. This consists of the triad of:
- Deficiency or absence of abdominal wall musculature.
- Bilateral cryptorchidism (in boys).
- Ureter, bladder and urethral abnormalities – VUR, megaureter, megacystits, PUJO, VUJO, urethral dilatation or atresia.
There is an incidence around 1 in 30 000 – 40 000 births, 95% are males. In 75% of cases there are other associated anomalies:
- GI e.g. malrotation, gastroschisis, imperforate anus.
- Skeletal e.g talipes, DDH.
- Cardiac
- Pulmonary (hypoplasia secondary to oligohydramnios).
Most cases are identified antenatally with severely dilated urinary tracts and distended abdomen. Postnatally detection is on examination with a wrinkled and lax appearance of the abdominal wall, palpable bladder, and cryptorchidism. Neonatal survival may depend on the degree of pulmonary hypoplasia, and presence of non-renal defects, listed above. There is a variable degree of renal dysplasia which makes chronic kidney disease common, exacerbated by recurrent UTI and poor urinary drainage or VUR.
Initial management will involve assessment of respiratory status and requirement for respiratory support where there is noted to be severe oligohydramnios antenatally. Subsequently, such infants will require investigation for known renal and non-renal anomalies. Long term renal outcomes are very guarded, severe dysplasia may be associated with early end stage renal failure but all will have a degree of impairment and abnormal drainage and bladder function. There are specific challenges offering renal replacement in these children.
It is most likely that cases identified antenatally will have a documented plan for immediate postnatal management. Otherwise immediate renal management will involve admission to NNU & urgent renal tract USS within 24-48 hours of birth and referral to the nephrology team within the same time frame. Many will require catheter drainage in the short term and vesicostomy as a more permanent drainage procedure. Daily monitoring of BP, renal function, urine output and fluid balance will be required pending further investigations.
Persistent Cloaca
|
Antenatal Counselling |
Refer to Neonatology & Urology Teams |
|
Attendance at Delivery/ Admission |
Not routinely indicated. Admission to NNU required. |
|
Imaging |
Urgent Renal USS 24-48 hours |
|
Referral |
Inform urology team after delivery |
|
Prophylaxis |
Commence antibiotic prophylaxis pending further imaging. |
|
Genetics |
Referral not indicated. |
|
Other Monitoring |
Daily BP, U&E, urine output and fluid balance required. |
Occurs in females where there is failed development of the urorectal septum causing the urethra, vagina and colon to open into a single channel. Can cause obstruction to any of the three tracts. Relief of obstruction is the management priority, followed by reconstructive surgery. Discuss with urology if suspected antenatally. Urgent urology referral if anatomical concerns postnatally.
Bladder Exstrophy
|
Antenatal Counselling |
Refer family to Neonatal and Urology teams |
|
Attendance at Delivery/ Admission |
Paediatrics to attend delivery. Admission to NNU required.Bladder plate to be covered with mepitel following admission. |
|
Imaging |
Urgent renal USS 24-48 hours. |
|
Referral |
Inform urology team after delivery. |
|
Prophylaxis |
Commence antibiotic prophylaxis pending further imaging. |
|
Genetics |
Referral not indicated. |
|
Other Monitoring |
Daily BP, U&E, urine output and fluid balance required. |
This occurs due to failure of the infraumbilical mesenchyme to separate the cloaca that will form the bladder from overlying ectoderm in fetal development. There is breakdown of cloacal membrane which causes exposure of the posterior bladder wall, shortened abdominal wall, incomplete fusion of genital tubercles, separated pubic rami and inguinal herniae. Baby has defective lower abdominal wall and multiple abnormalities of pelvis, bladder, urethra and external genitalia. Incidence is approximately 2 per 100000 live births, Male:Female ratio is 6:1. Most are identified antenatally and will have been discussed with specialist teams prior to delivery. There should be a postnatal management plan in place. Management requires complex surgery which is only performed in GOSH or Manchester.
Compensatory hypertrophy of the remaining solitary kidney can be defined as growth above the 50th centile (length) for age and height. Alternatively, Rottenberg et al produced normograms for kidney length, by age in children with a single kidney (see table below).
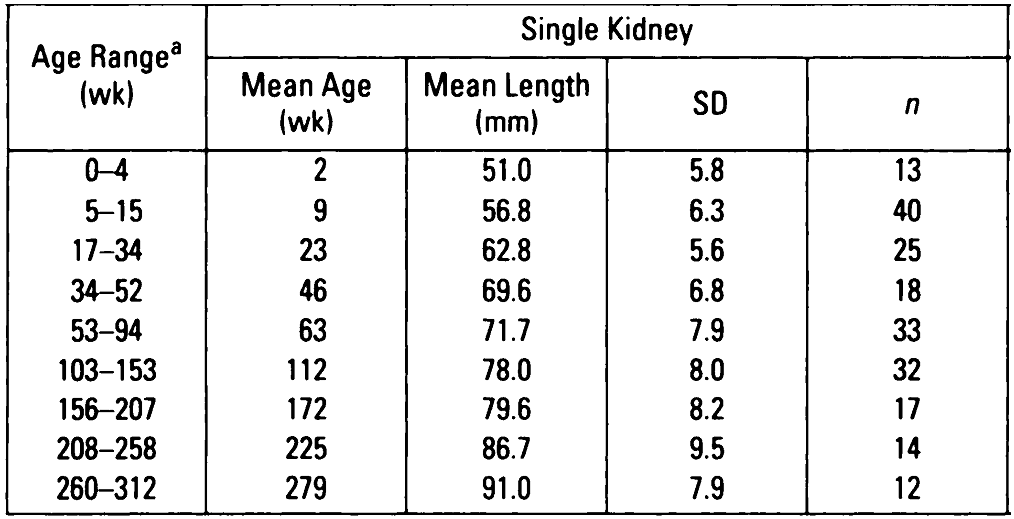 Rottenberg GT; De Bruyn R; Gordon I. Sonographic standards for a single functioning kidney in children. American Journal of Roentgenology. 167(5):1255-9.
Rottenberg GT; De Bruyn R; Gordon I. Sonographic standards for a single functioning kidney in children. American Journal of Roentgenology. 167(5):1255-9.
Adiego B, Martinez-Ten P, Perez-Pedregosa J, Illescas T, Barron E, Wong AE, Sepulveda W. Antenatally diagnosed renal duplex anomalies: sonographic features and long-term postnatal outcome. J Ultrasound Med. 2011 Jun;30(6):809-15
Blazer S, Zimmer EZ, Blumenfeld Z, Zelikovic I, Bronshtein M. Natural history of fetal simple renal cysts detected in early pregnancy. J Urol. 1999 Sep;162(3 Pt 1):812-4.
Capisonda R, Phan V, Traubuci J, Daneman A, Balfe JW, Guay-Woodford LM. Autosomal recessive polycystic kidney disease: outcomes from a single-center experience. Pediatr Nephrol. 2003 Feb;18(2):119-26
Castagnetti M, Cimador M, Esposito C, Rigamonti W. Antibiotic prophylaxis in antenatal nonrefluxing hydronephrosis, megaureter and ureterocele. Nat Rev Urol. 2012 May 8;9(6):321-9
Chaumoitre K, Brun M, Cassart M, Maugey-Laulom B, Eurin D, Didier F, Avni EF. Differential diagnosis of fetal hyperechogenic cystic kidneys unrelated to renal tract anomalies: A multicenter study. Ultrasound Obstet Gynecol. 2006 Dec;28(7):911-7.
Damen-Elias HA, De Jong TP, Stigter RH, Visser GH, Stoutenbeek PH. Congenital renal tract anomalies: outcome and follow-up of 402 cases detected antenatally between 1986 and 2001. Ultrasound Obstet Gynecol. 2005 Feb;25(2):134-43.
Deshpande C, Hennekam RC. Genetic syndromes and prenatally detected renal anomalies. Semin Fetal Neonatal Med. 2008 Jun;13(3):171-80.
Estrada CR Jr, Passerotti CC, Graham DA, Peters CA, Bauer SB, Diamond DA, Cilento BG Jr, Borer JG, Cendron M, Nelson CP, Lee RS, Zhou J, Retik AB, Nguyen HT. Nomograms for predicting annual resolution rate of primary vesicoureteral reflux: results from 2,462 children. J Urol. 2009 Oct;182(4):1535-41.
Fanos V, Cataldi L. Antibiotics or surgery for vesicoureteric reflux in children.Lancet. 2004 Nov 6-12;364(9446):1720-2
Kwatra N, Shalaby-Rana E, Majd M. Scintigraphic features of duplex kidneys on DMSA renal cortical scans. Pediatr Radiol. 2013 Sep;43(9):1204-12
Mashiach R, Davidovits M, Eisenstein B, Kidron D, Kovo M, Shalev J, Merlob P, Vardimon D, Efrat Z, Meizner I. Fetal hyperechogenic kidney with normal amniotic fluid volume: a diagnostic dilemma. Prenat Diagn. 2005 Jul;25(7):553-8.
Mathews R, Carpenter M, Chesney R, Hoberman A, Keren R, Mattoo T, Moxey-Mims M, Nyberg L, Greenfield S. Controversies in the management of vesicoureteral reflux: the rationale for the RIVUR study. J Pediatr Urol. 2009 Oct;5(5):336-41
Mattoo TK. Diagnosis of congenital renal anomalies in children.Clin Biochem. 2011 May;44(7):498
Munding M, Al-Uzri A, Gralnek D, Riden D. Prenatally diagnosed autosomal recessive polycystic kidney disease: initial postnatal management. Urology. 1999 Dec;54(6):1097
Onal B, Kogan BA. Natural history of patients with multicystic dysplastic kidney-what followup is needed? J Urol. 2006 Oct;176(4 Pt 1):1607-11.
Sanna-Cherchi S, Ravani P, Corbani V, Parodi S, Haupt R, Piaggio G, Innocenti ML, Somenzi D, Trivelli A, Caridi G, Izzi C, Scolari F, Mattioli G, Allegri L, Ghiggeri GM. Renal outcome in patients with congenital anomalies of the kidney and urinary tract. Kidney Int. 2009 Sep;76(5):528-33
Skoog SJ, Peters CA, Arant BS Jr, et al. Pediatric Vesicoureteral Reflux Guidelines Panel Summary Report: Clinical Practice Guidelines for Screening Siblings of Children With Vesicoureteral Reflux and Neonates/Infants With Prenatal Hydronephrosis.J Urol. 2010 Sep;184(3):1145-51.
Smellie JM, Barratt TM, Chantler C, Gordon I, Prescod NP, Ransley PG, Woolf AS. Medical versus surgical treatment in children with severe bilateral vesicoureteric reflux and bilateral nephropathy: a randomised trial. Lancet. 2001 Apr 28;357(9265):1329-33.
Tekgül S, Riedmiller H, Hoebeke P, Kočvara R, Nijman RJ, Radmayr C, Stein R, Dogan HS; European Association of Urology. EAU guidelines on vesicoureteral reflux in children. Eur Urol. 2012 Sep;62(3):534-42.
Tsatsaris V, Gagnadoux MF, Aubry MC, Gubler MC, Dumez Y, Dommergues M. Prenatal diagnosis of bilateral isolated fetal hyperechogenic kidneys. Is it possible to predict long term outcome? BJOG. 2002 Dec;109(12):1388-93.
van den Bosch CM, van Wijk JA, Beckers GM, van der Horst HJ, Schreuder MF, Bökenkamp A. Urological and nephrological findings of renal ectopia. J Urol. 2010 Apr;183(4):1574-8.
Westland R, Schreuder MF, Ket JC, van Wijk JA. Unilateral renal agenesis: a systematic review on associated anomalies and renal injury. Nephrol Dial Transplant. 2013 Jul;28(7):1844-55.
Whitten SM, McHoney M, Wilcox DT, New S, Chitty LS. Accuracy of antenatal fetal ultrasound in the diagnosis of duplex kidneys. Ultrasound Obstet Gynecol. 2003 Apr;21(4):342-6.
Wiesel A, Queisser-Luft A, Clementi M, Bianca S, Stoll C; EUROSCAN Study Group. Prenatal detection of congenital renal malformations by fetal ultrasonographic examination: an analysis of 709,030 births in 12 European countries. Eur J Med Genet. 2005 Apr-Jun;48(2):131-44.
Yohannes P, Smith AD. The endourological management of complications associated with horseshoe kidney. J Urol. 2002 Jul;168(1):5-8.
Last reviewed: 09 August 2022
Next review: 31 August 2023
Author(s): Dr Andrew Cooper, Consultant Neonatologist - RHC; Dr C Lilley – Neonatal consultant – Princess Royal Maternity (RPD guideline); Dr Rosalyn Ardill – Consultant Nephrologist - RHSC, Edinburgh
Version: 2
Co-Author(s): Other professionals consulted: Dr David Hughes - Consultant Paediatric Nephrologist - RHC; Mr Stuart O’Toole – Consultant Paediatric Urologist – RHC; Dr Carol Gardiner – Consultant Geneticist – RHC; Mr Chris Driver – Consultant Urologist, ACH, Aberdeen; Dr Andrew Watt – Consultant Radiologist, RHC
Approved By: West of Scotland Neonatology Guideline Group
Document Id: 949