
Guideline on the classification and management of glomerulonephritis in general Paediatrics
exp date isn't null, but text field is
Scope
Patients diagnosed or being investigated for glomerulonephritis.
Audience
Medical and nursing staff managing children with renal disease.
Glomerulonephritis (GN) is a broad term used to describe several diseases which cause glomerular inflammation and injury. The kidneys are involved symmetrically and the cause may be specific to the renal system (primary cause) or part of a systemic disease (systemic cause). The different clinical presentations of glomerulonephritis include:
- acute GN – sudden onset haematuria and proteinuria +/- oedema +/- hypertension with normal or mildly impaired renal function
- rapidly progressing GN– as above with declining renal function over days
- recurrent episodes – recurrent macroscopic haematuria usually preceded by a viral URTI
- chronic GN – haematuria and proteinuria, depending on stage of disease often with relatively static chronic renal impairment +/- hypertension.
Each clinical presentation could be one of several different renal diseases. Diagnosis is based upon the presentation, presence of extra-renal findings, family history, blood and urine testing and in some cases histology from a renal biopsy.
This guideline aims to;
- discuss the typical presentation, differential diagnosis and management of acute GN
- discuss criteria for referral to/discussion with the nephrology team
- provide an overview of each differential diagnosis and basis of management
- provide an overview of rapidly progressive glomerulonephritis
Acute glomerulonephritis (AGN) is a syndrome consisting of frank haematuria, proteinuria, oliguria, volume overload and usually a mild increase in plasma creatinine. The differential diagnosis of AGN shown in box 1. Although nephritic and nephrotic syndromes are often discussed as separate entities they can exist together. Any proteinuria which is in nephrotic range (Protein:creatinine ratio >200mg/mmol) may cause a nephrotic syndrome.
Box 1
|
Common causes
Less common causes
Rare causes
|
* most commonly presents as a nephrotic syndrome – see section 8.
2.1 Initial Investigations of AGN
A detailed history and examination is, as always, important. Areas for particular emphasis are shown in box 2.
Box 2
|
Blood: FBC, U&E, LFTs, Calcium and phosphate, Immunoglobulins, ASO titre/antiDNAse B titres, Complement (abnormalities shown in box 3), ANA, ANCA, Anti GBM
Urine: Urine protein creatinine ratio (PCR), Culture
Imaging: Renal USS
Box 3
|
Complement abnormalities in glomerulonephritis* Normal C3 & C4
Decreased C3 &Normal C4**
Decreased C3 & C4
|
* Some conditions may give more than one complement pattern
** If C3 low and post infectious GN suspected, complement must be checked again in 6-8 weeks. If still low – may not be post infectious.
2.2 Management of AGN
The initial management of AGN is largely supportive with focus on fluid and salt restriction and management of hypertension. The definitive management is guided by the underlying diagnosis and will be discussed in each section below. Suggested monitoring of inpatients is shown in box 4 below.
Box 4
|
The aim of fluid management in GN is to match input with output – ie input should be approximately equal to urine output plus insensible losses (400ml/m2/24 hours). This will involve regular assessment of the fluid balance chart to ensure urine output is not decreasing.
Hypertension is usually due to fluid overload and often responds to diuretics; however, a calcium channel blocker may also be useful, particularly if the patient is ready for discharge home and still hypertensive. Suggested starting doses are indicated below:
- Furosemide 0.5mg/kg/dose once daily. It can be titrated to 1mg/kg/dose twice daily.
- Amlodipine 0.1mg/kg/dose once daily. It can be incremented up to 0.4mg/kg/dose at 48 hour intervals if no effect.
- Nifedipine 250-500 micrograms/kg (max 20mg) 8 hourly. It has a much shorter half-life than Amlodipine so can be titrated to effect more quickly. It is only suitable in older children who can take tablets.
2.3. Indications for referral
Any patients with complications in the acute phase of the illness such as those listed in box 5 should be urgently discussed with the renal team. Other indications for referral are discussed as ‘non-urgent discussion’ and ‘letter/email referral’ and are listed in boxes 6 and 72.
Box 5 – Indications for immediate discussion
|
Box 6 – Indications for non-urgent discussion
|
Box 7 – Letter/email referral
|
The most common aetiological factor of paediatric GN is prior infection (80% of cases), with group A beta haemolytic streptococcus being the most common causative organism. The acute nephritis typically occurs 1-2 weeks after a history of a pharyngeal streptococcal infection, or less commonly 3-6 weeks after a skin infection. It commonly occurs between the ages of 2 – 15 with median age of onset 6-8 years1.
In prior streptococcal infection ASO titre will be elevated. Typically C3 is low and C4 is normal. C3 will usually return to normal in 6-8 weeks and if low should be checked again after this time. If it has not returned to normal then other causes such as SLE or MPGN should be considered. Figure 1 shows a typical pattern of C3, ASOT, hypertension and haematuria following group A strep infection.
Figure 12
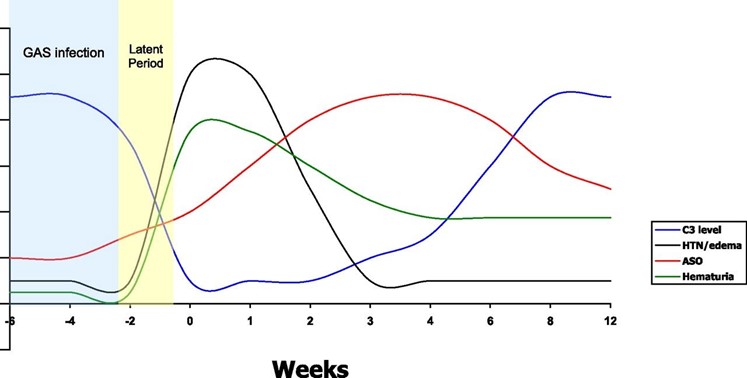
The use of Penicillin has little effect on the natural history of the disease but it can be useful in eradicating nephritic strains of group A streptococcus. A 10 day course is therefore indicated.
Severity and duration of post streptococcal GN is variable but most clinical symptoms resolve within 2-3 weeks. Microscopic haematuria alone may exist for 1-2 years and is of no long term relevance3.
Primary IgA can occur at all ages but is most common during the second or third decades of life and is more common in males. Patients with IgA nephropathy typically present in one of three ways;
- Recurrent macroscopic haematuria
- Asymptomatic microscopic haematuria & proteinuria
- Acute nephritic syndrome and/or nephrotic syndrome
A typical presentation of primary IgA is recurrent macroscopic haematuria occurring 2-3 days after the onset of an upper respiratory tract infection. The interval between precipitating infection and the onset of haematuria is much shorter than the 1-2 weeks seen in post infectious GN. Diagnosis is largely a clinical one and renal biopsy for a definitive diagnosis is not indicated in most patients. The outcome of this condition is also varied but presentation with an acute nephritic and/or nephrotic syndrome and degree of proteinuria is thought to correlate to the severity of the glomerular injury. Amongst patients who develop overt proteinuria and/or an elevated serum creatinine, the progression to end stage renal failure is 15 - 20% at 10 years and 20-30% at 30 years4.
There are 2 approaches to the treatment of IgA nephropathy.
- General interventions to slow progression to end stage renal failure that are non-specific to IgA. These include blood pressure control and ACE inhibitor use in patients with proteinuria.
- Immunotherapy with glucocorticoids +/- other immunosuppression to treat underlying inflammatory disease.
As there are no serological markers to measure disease activity, clinical parameters such as serum creatinine, urinary PCR, blood pressure and estimated GFR should be used. With all this in mind treatment recommendation is:
- PCR 20-100mg/mmol – observation and 3 monthly assessments if PCR appears stable.
- PCR 100-200mg/mmol - ACE inhibitor.
- PCR > 200mg/mmol after maximal ACE inhibitor therapy for 3 – 6 months should be considered for glucocorticoid therapy
Other considerations for glucocorticoid therapy are a progressively declining GFR and active disease on biopsy. Glucocorticoids are not indicated in patients with chronically elevated serum creatinine or chronic changes on biopsy.
HSP is a small vessel vasculitis. It is relatively common with an incidence of 14 per 100,000 children and its peak is around 4-5 years of age with a male predominance. The diagnosis is a clinical one. Typically the disease is characterised by involvement of 4 organs. The skin (symmetrical purpuric rash over lower limbs and buttocks), the gastrointestinal tract (ranging from mild abdominal pain to malena and intussusception), joints (arthralgia or arthritis) and the kidneys.
Renal involvement occurs in 20-54% of all patients with HSP and it is more common in older children (renal involvement refers to haematuria, proteinuria, hypertension and reduced GFR)6. The extent of renal involvement is variable ranging from transient mild haematuria/proteinuria to a severe nephritic picture. Nephrotic range proteinuria, hypertension and the coexistence of haematuria and proteinuria are associated with an increased risk of progressive disease. The renal management for HSP is as per flow diagram in the HSP guideline.
Biopsy findings are identical to those found in IgA nephropathy. The Oxford classification for IgA nephropathy has recently changed and now involves the MEST criteria which are independent prognostic factors and are highlighted in box 8. The presence of endocapillary hypercellularity (see box 8) indicates likely response to immunosuppressive treatment.
Box 8 – MEST classification of IgA nephropathy11
0 = <50% 1= >50%
0 = Absent 1= Present
0 = Absent 1= Present
0 = Absent or <25% of cortex 1 = 25-50% of cortex 2 = >50% of cortex |
Specific treatment for HSP nephritis should be considered in patients with marked proteinuria and/or impaired renal function during the acute episode. A renal biopsy should be obtained prior to initiating treatment. As outlined above, the severity of the histological findings is the best indicator of prognosis and will direct treatment.
An ACE inhibitor is indicated in patients with proteinuria. In patients with severe disease, immunosuppression with high-dose Methylprednisolone 600mg/m2 for 3 days followed by oral Prednisolone 1mg/kg/day for 3 months is indicated. Cyclophosphamide is still used but there is little evidence for it. There is some evidence that Ciclosporin may be of use6.
SLE is a chronic inflammatory multisystem disease of unknown cause. Serum positivity for ANA is present in almost all children with SLE. Anti-double stranded DNA may help to confirm the diagnosis but is not uniformly present. Renal involvement is relatively common with approximately 60% of children having some involvement at presentation9. Proteinuria is the most frequently observed abnormality although any feature of nephritis can occur. Clinical findings often underestimate the extent of renal involvement and some patients may have significant pathological findings on biopsy with minimal clinical signs.
Treatment of acute nephritis is as outlined above and if a diagnosis of SLE is suspected, a multidisciplinary approach, lead by the rheumatology team is preferable.
The histopathological classification of lupus nephritis is important in consideration of treatment and is shown in box 9 below.
Box 9 – Histopathological classification of lupus10
|
Immunosuppressive therapy is indicated in patients with diffuse or focal proliferative disease on renal biopsy (ie class III and above). This includes intravenous Methylprednisolone in the acute phase followed by oral Prednisolone and consideration of Cyclophosphamide, Azathioprine and/or Mycophenolate mofetil (MMF).
MPGN is the name given to a pattern of glomerular injury viewed by light microscopy. It is predominantly a disease of older children and young adults with boys and girls equally affected. It may present initially as haematuria and/or proteinuria or as an acute nephritic or nephrotic syndrome3. There are several potential causes, the classification of which has changed in recent years. It should be considered in patients who present with glomerulonephritis who have a persistently low complement levels or in steroid resistant nephrotic syndrome. Such patients should be referred for consideration of a biopsy. The management of MPGN is discussed in a separate guideline.
FSGS is a histological diagnosis rather than a disease and is based on the presence of focal and glomerular segmental scarring on biopsy. It typically presents as a steroid resistant nephrotic syndrome but it can also present with asymptomatic proteinuria and/or haematuria.
FSGS may be classified according to the known or presumed cause:
- Primary or idiopathic (without an identifiable cause)
- Secondary (in response to previous glomerular injury)
- Other causes such as infections, toxins and genetic abnormalities.
Primary FSGS typically presents as a nephrotic syndrome whilst secondary typically presents with non-nephrotic range proteinuria and often renal insufficiency. Making this distinction is not always easy but it is important as it can effect which treatment is chosen. Primary FSGS is more likely to respond to immunosuppressive therapy.
Often patients with FSGS will have had at least 4 weeks of oral Prednisolone and consideration of IV Methylprednisolone prior to a renal biopsy (in the treatment of their nephrotic syndrome). Thereafter, there are a range of treatment options which will be considered on an individual case basis. Possible treatment includes Tacrolimus, MMF, Ciclosporin as well as plasmapheresis and Rituximab.
Children presenting with glomerulonephritis accompanied by rapid (days/weeks) loss of renal function are said to have RPGN. This clinical picture is usually associated with crescentic glomerulonephritis on renal biopsy. This is caused by increased permeability of the glomerular basement membrane (GBM) leading to layers of circulating T cells, macrophages, fibroblast and epithelial cells in the Bowman’s space. There are a number of diseases that can cause this.
The mechanisms of the diseases that can cause RPGN are divided into 3 broad categories:
- Immune complex
- Pauci-immune
- Anti-Glomerular basement membrane (GBM) disease
As the name suggests, immune complex mediated RPGN refers to disease with the presence of immune deposits in the glomeruli. This includes post infectious GN, HSP, SLE and MPGN and is the most commonly observed pattern. Pauci-immune refers to a vasculitis with little or no immune deposits seen on biopsy. These conditions include granulomatosis with polyangitis, microscopic polyangitis and Churg-Struass syndrome and are typically ANCA positive. Anti-GBM disease is typified by linear GBM staining and measurable levels of circulating anti-GBM antibody. Goodpastures disease is the name given to the combination of glomerulonephritis with pulmonary haemorrhage in the presence of circulating anti-GBM antibodies.
Consideration of the underlying cause, particularly in immune complex mediated RPGN will direct treatment. It is likely that the treatment regimen will involve high dose IV Methylprednisolone (600mg/m2) for 3 days followed by an oral weaning regimen over 6 months. Anti GBM disease will involve Cyclophosphamide and consideration of plasma exchange. These treatments would also be considered in pauci-immune disease if dialysis dependent at the time of presentation.
Most cases of RPGN will progress to end stage renal failure requiring dialysis and transplantation but early recognition and referral for treatment may help to minimise the degree of reversible renal injury.
- Eison TM, Ault BH, Jones DP, Chesney RW, Wyatt RJ. (2011) Post-streptococcal acute glomerulonephritis in children: clinical features and pathogenesis. Paediatric Nephrology; 26: 165 – 180
- Vandeevoorde RG. Acute post streptococcal glomerulonephritis: The most common acute glomerulonephritis. Paediatrics in review, January 2015, Vol 36/Issue 1.
- Rees L, Brogan PA, Brockenhauer D, Webb NJA. Paediatric nephrology. Second edition. Oxford University press 2012.
- Barrat J, Freehally J. Clinical presentation and diagnosis of IgA nephropathy. Up to date. September 2018
- Geary DF, Schaefer F. Comprehensive Paediatric nephrology. Mosby Elsevier 2008
- Dedoeglu F, Kim S. Henoch Schonlein Purpura (immunoglobulin A vasculitis): Clinical manifestations and diagnosis. Up to date. December 2015.
- Fervenza FC, Sethi S. Evaluation and treatment of membranoproliferative glomerulonephritis. Up to date. September 2018
- Cattran DC, Appel GB. Treatment and prognosis of IgA nephropathy. Up to date. September 2018
- Thomas JA, Lehman MD. Systemic lupus erythromatosis in children: Clinical manifestations and diagnosis. Up to date. August 2016.
- Weening JJ et al. On behalf of the International society of nephrology and renal pathology society working group on the classification of lupus nephritis. The classification of glomerulonephritis in systemic lupus erythematosus revisited. Journal of the American society of Nephrology 15:241-250 2004.
- Roberts, IA. Oxford classification of immunoglobulin A nephropathy: an update. Current opinion in nephrology and hypertension. 2013 22:281 – 286.
Last reviewed: 30 September 2018
Next review: 06 January 2024
Author(s): Sheena Logan
Co-Author(s): Deepa Athavale, Consultant Paediatric Nephrologist
Approved By: Paediatric Clinical Effectiveness & Risk Committee