
Haemophilia protocol
exp date isn't null, but text field is
Objectives
The Royal Hospital for Children is the regional centre (West of Scotland) for the management of children with inherited bleeding disorders. The purpose of this protocol is to provide information of the appropriate use of blood products for this patient group.
Scope
Children with haemophilia and other clotting factor deficiencies.
Audience
Clinicians involved in the treatment of children with these conditions, including acute bleeding and surgery.
This SOP will cover:
-
Products used for the management of inherited bleeding disorders (5.1)
-
Management of IBDs – general points (5.2)
-
Treatment of Haemophilia A (5.3)
-
Treatment of Haemophilia A Patients with a FVIII Inhibitor
-
Laboratory monitoring in patients on Emicizumab (Hemlibra)
-
Treatment of Haemophilia B (5.4)
-
Treatment of von Willebrand’s Disease (5.5)
-
Factor XIII Deficiency (5.6)
-
Factor VII Deficiency (5.7)
-
Factor XI Deficiency (5.8)
-
Specific Bleeding Situations (5.9)
-
Products – Sourced from Blood Bank via Trakcare (5.10)
-
UKHCDO Maintenance Dose Table for Emicizumab (Appendix 1)
-
How to Order Factor Replacement Products from Trakcare (Appendix 3)
-
Blood Product Collection (Appendix 4)
None.
3.1 The management of haemophilia patients will be directed by the Consultant or a senior member of the medical team.
3.2 the Medical/Nursing team will be responsible for admitting, assessing, investigating and administering treatment, and monitoring response.
None.
1. Factor Concentrate:
- Standard half life (SHL) FVIII & FIX : several products are available - see individual disorders & products
- Extended half-life (EHL) FVIII and FIX products are allocated for some patients
- Other specific factor concentrates e.g. vWF containing products, FXIII – see individual disorders & products.
2. Emicizumab (Hemlibra)
This is a bi-specific monoclonal antibody used for prophylaxis in Haemophilia A (in patients with & without inhibitors).
3. DDAVP (Desmopressin):
Used for some children with mild haemophilia or von Willebrand disease. This will usually be indicated in a care plan with a record of response (DDAVP trial).
4. Anti-fibrinolytic Agents (Tranexamic Acid):
- Orally 25mg/kg 2-3 times per day (max dose 1.5G)
- Intravenously 10mg/kg 2-3 times per day (max dose 1G)
Can be used alone or with factor concentrate/DDAVP
Most useful for mucosal bleeding
Contraindicated in the presence of haematuria
General Information:
- All children should have a care plan on clinical portal which will provide information on baseline levels and allocated treatment products to use in the presence of bleeding problems. Recent clinic letters may also be helpful
- Some children with severe disorders are treated via ports (or hickman lines). Children presenting with fever should be assessed for a potential line infection/sepsis and admitted for antibiotics via the haematology service.
- NSAIDs are generally contraindicated for children with inherited bleeding disorders and should only be administered following discussion with a consultant.
- All significant injuries/major bleeds; bleeds that do not settle and any surgical interventions should be discussed with a Consultant.
Most children with severe haemophilia A will be on regular prophylaxis either with FVIII (standard half life or extended half life product) or with Emicizumab.
Children with moderate and mild haemophilia are usually treated “on demand” i.e. in the presence of bleeding or following trauma.
Emicizumab (Hemlibra) use in Severe Haemophilia A:
Emicizumab (Hemlibra) is a bispecific antibody used for prophylaxis in severe haemophilia A. It can be used in both inhibitor and non-inhibitor patients. It is administered by subcutaneous injection, either weekly or every 2 weeks (see Appendix 1for dosing information).
Emicizumab is only used for prophylaxis and is not used for management of bleeding episodes. Treatment of bleeds will depend on whether the patient has an inhibitor or not.
Management of bleeds in patients on Emicizumab prophylaxis who do not have an inhibitor:
The use of tranexamic acid should be considered in all bleeding episodes and may be adequate for minor bleeds/trauma.
In patients without inhibitors who require FVIII therapy bleeds should be managed as per standard guidance. Care should however be taken to avoid high post treatment levels (>150iu/dl).
All patients on emicizumab prophylaxis are allocated to a standard half life product for the management of bleeding.
See section ‘Severe/Moderate Haemophilia A’ below.
Severe/Moderate Haemophilia A:
FVIII replacement with allocated product, ordered via Trakcare (see Appendix 3)
FVIII dosing guideline for standard half life products:
Factor VIII increase is 2 iu/dl for every iu/kg of FVIII infused.
Required units = body weight (kg) x desired factor VIII:
C rise (iu/dl) x 0.5
(can be less in young children)
Table 1:
|
Bleed Site (target) |
Haemophilia A |
|
Minor injuries/accidents; (target 40iu/dl) |
20iu/Kg |
|
Moderate injuries/accidents; |
30iu/Kg |
|
Major bleeds; |
40-50iu/Kg |
Laboratory monitoring in patients on emicizumab:
Emicizumab affects routine standard bloods tests used to monitor FVIII replacement and tests used to assess inhibitors. Specific tests are therefore required.
For monitoring FVIII replacement in the presence of emicizumab use a chromogenic FVIII assay (Track request – see appendix 2). Note this assay is performed at GRI and out of hours urgent tests will have to be discussed with the on call consultant at GRI.
For measuring inhibitor levels request a chromogenic inhibitor assay (Track request – see appendix 2). Note this assay is performed at GRI.
On routine coagulation screening in patients on emicizumab the APTT should be shortened (or normal). Prolongation of the APTT may suggest the presence of an anti-drug antibody (ADA) against emicizumab. As this may affect efficacy further assessment will be required either using an emizicumab assay or a specific anti-drug antibody assay. This should be discussed with a consultant and/or the haemostasis lab.
Management of bleeds in patients on FVIII prophylaxis:
Patients may be on either a standard half life or and extended half life product for prophylaxis.
In general this does not affect the initial dose given but may affect the scheduling of follow up doses.
For initial FVIII dosing see Table 1 above.
The use of tranexamic acid should be considered for minor bleeding, particularly mucosal bleeding and can also be use together with FVIII.
Management of bleeding in patients on extended half life FVIII products (e.g. Elocta)
First infusion should raise FVIII levels appropriate to the bleed as above
Subsequent dosing with EHL products should take into account previous half life studies (see Trakcare care plan & discuss with consultant on call as required) and additional monitoring may be required.
Monitoring of FVIII replacement:
Factor VIII levels should be monitored in the presence of severe bleeding and/or following surgery, (pre dose administration and 15 mins post dose, using coag bottle)
Management of Mild Haemophilia A:
FVIII replacement with allocated product, ordered via Trakcare (see appendix 2)
OR
DDAVP - (where a response has been demonstrated)
NOTE
Children <2yrs should not receive DDAVP, because of the risk of fluid overload and hyponatraemia. Use allocated factor concentrate.
Children 2-3yrs who require treatment with DDAVP, should be admitted and have their electrolyte and fluid balance monitored for at least 24 hours following DDAVP. Excess fluid intake should be avoided.
FVIII dosing guideline
For patients requiring FVIII calculate dose based on the baseline FVIII level and target FVIII.
Factor VIII increase is 2 iu/dl for every iu/kg of FVIII infused
Required units = body weight (kg) x desired factor VIII:
C rise (iu/dl) x 0.5
DDAVP dosing guideline
DDAVP should result in a 3-5 fold increase from baseline FVIII
For individual DDAVP response see care plan.
Vials contain 4mcg/ml OR 15mcg/ml (Octim)
- Dose 0.3mcg/kg to a maximum dose of 20mcg/dose
- Administration - subcutaneously or intravenously.
- Intravenous administration- prescribed amount is added to 50mls 0.9% NaCl and given intravenously over 45-60 mins via butterfly or cannula.
- Subcutaneous injection - give required amount.
If DDAVP is required on two or more consecutive days, U&E’s and fluid balance should be monitored.
Factor VIII levels should be monitored in the presence of severe bleeding and/or following surgery, (pre dose administration and 15 mins post dose, using coag bottle)
Guidance for Mild Haemophilia A:
DDAVP 0.3mcg/KG (Max dose to be given 20mcg/dose)
|
Bleed Site (target) |
Haemophilia A |
|
Minor injuries/accidents; (target 40-50iu/dl) |
0.3mcg/kg either IV or |
| Moderate injuries/accidents; (target 60-80iu/dl) major muscle bleeds; joint bleeds; long bone #; Haematuria |
0.3mcg/kg either IV or |
|
Major bleeds; (target 80-100iu) |
Use allocated factor product |
Management of bleeding in haemophilia A patients with inhibitors:
Patients with inhibitors are likely to respond poorly to standard doses of FVIII.
They may be receiving regular FVIII as part of an immune tolerance regimen.
They may be receiving prophylaxis with emicizumab.
Options for the treatment of bleeds are:
- High dose FVIII or with r-FVIIa (occasionally FEIBA).
- Which treatment to use will be detailed as part of the patients care plan along with recommended dosing.
- When using FVIII in patients on emicizumab prophylaxis care should be taken to avoid high post treatment levels (>150iu/dl).
- Thrombotic events have been reported with r-FVIIa and it may be possible to manage bleeds with a low dose regimen. See below.
Feiba is occasionally required for the management of bleeds in inhibitor patients but in combination with emicizumab has been associated with thrombosis and TMAs. It should therefore be used with extreme caution (in accordance with national guidance) and only as specifically directed by a consultant.
Dosing Novoseven (rFVIIa)
Choice of dose will depend on the nature of the bleed and whether the patient is receiving emicizumab for prophylaxis.
See care plan for individual patient guidance.
1mg, 2mg and 5mg vials are available, doses should be rounded to the nearest whole vial size, where appropriate.
Patients with inhibitors not on Emicizumab prophylaxis.
Mild-moderate bleeding/injuries
- 2-3 injections of 90 mcg per kg administered at 2 hourly intervals
- If further treatment is required, one additional dose of 90 mcg per kg can be administered
Haemarthrosis/significant bleeds/injuries
- One single injection of 270 mcg per kg body weight (NB high dose r-FVIIa is contraindicated in children on emizicumab who should only receive standard dosing – see below & individual care plans)
- Subsequent dosing will vary according to the type and severity of bleeding.
Major/unresolved bleeding episodes
- 90 mcg per kg every 2 hours and should continue until clinical improvement is observed. Dosing can then be extended to 3hourly for 24 hrs, 4hourly for 24 hours.
Invasive procedure/surgery
- An initial dose of 90 or 270 mcg/kg should be given immediately before the intervention.
- 2 hourly dosing of 90mcg/kg should then proceed for the first 24 - 48 hours depending on the intervention performed and the clinical status of the patient.
Patients with inhibitors on Emicizumab prophylaxis.
NB avoid high dose rFVIIa in patients on emicizumab.
Mild-moderate bleeding/injuries
- Starting dose of 45mcg/kg
- If poor response dose may be increased to 90mcg/kg
- Subsequent dosing will vary according to the type and severity of bleeding.
Haemarthrosis/significant bleeds/injuries
- Starting dose of 90mcg/kg (NB high dose r-FVIIa is contraindicated in children on emizicumab who should only receive standard dosing – see below & individual care plans)
- Subsequent dosing will vary according to the type and severity of bleeding.
Invasive procedures/surgery
- For patients on emicizumab it may be possible to manage some surgery with tranexamic acid alone.
- Otherwise rFVIIa will be required. NB avoid high dose rFVIIa.
For elective surgery a surgical care plan should be available.
Emergency surgery should be discussed with the on call consultant.
Product: Recombinant FIX as per allocated product
FIX Dosing Guidelines:
Factor IX increase is approximately 1 iu/dl for every 1 iu/kg FIX infused
(often less in young children)
Required units = body weight (kg) x desired factor IX rise (iu/dl)
Guidance for Patients with Severe/Moderate Haemophilia B
|
Bleed Site (target) |
Haemophilia B |
|
Minor injuries/accidents; |
40iu/Kg |
|
Moderate injuries/accidents; |
60iu/Kg |
|
Major bleeds; |
80-100iu/Kg |
Factor IX levels should be monitored in the presence of severe bleeding and/or following surgery, (pre dose administration and 15 mins post dose, using coag bottle)
All significant injuries/major bleeds; bleeds that do not settle and any surgical interventions should be discussed with the Consultant in Charge or on-call Consultant Haematologist
Extended half life FIX (Alprolix, Idelvion)
First infusion should raise FIX levels appropriate to the bleed as above.
Subsequent dosing with FIX EHL products should take into account previous half life studies (see Trakcare care plan & discuss with consultant as required)
All significant injuries/major bleeds; bleeds that do not settle and any surgical interventions should be discussed with the Consultant in Charge or on-call Consultant Haematologist
Guidance for Mild Haemophilia B
- Calculate dose based on the baseline FIX level and target FIX
Side Effects of FIX Administration
- Anaphylaxis – which can be an indication of inhibitor development
Type I vWD:
- DDAVP - where a response has been demonstrated
(see previous dosing guidelines)
Type II vWD:
- DDAVP - where a response has been demonstrated
- Patients <2yrs should not be given DDAVP, because of the associated risk of fluid overload and hyponatraemia. These patients should receive
- FVIII/vWF Voncento.
- Patients 2-3yrs who require treatment with DDAVP (where a response has been demonstrated), should be admitted and have their electrolyte and fluid balance monitored for at least 24 hours following DDAVP. Excess fluid intake should be avoided.
Type IIB vWD:
- Voncento (pdFVIII/vWF) (DDAVP is usually contraindicated in this sub-type of vWD)
Type III von Willebrand’s Disease:
- Voncento (pdFVIII/vWF)
Voncento (pdFVIII/vWF) dosing for Type I, II & III vWD
Dosing will depend on the baseline FVIII & vWF levels and the desired increase (see Trakcare for baseline levels)
Generally, 1 IU/kg VWF:RCo raises the circulating level of VWF:RCo by 0.02 IU/ml (2 %).
Levels of VWF:RCo of > 0.6 IU/ml (60 %) and of FVIII:C of > 0.4 IU/ml (40 %) should be achieved
On-demand treatment
Usually 40 - 80 IU/kg of von Willebrand factor (VWF:RCo) corresponding to 20 - 40 IU FVIII:C/kg of body weight (BW) are recommended to achieve haemostasis.
An initial dose of 80 IU/kg VWF:RCo may be required, especially in patients with type 3 VWD where maintenance of adequate levels may require greater doses than in other types of VWD.
Repeat dosing may be administered every 12-24 hours.
Voncento should be clearly prescribed in vWF:RCo units (or state both vWF and FVIII doses on the Kardex).(vials state both FVIII and vWF levels e.g. FVIII 500iu/vWF 1200).
FVIII & vWF activity levels should be monitored in the presence of severe bleeding and/or following surgery, (pre dose administration and 15 mins post dose, using coag sample).
All significant injuries/major bleeds; bleeds that do not settle and any surgical interventions should be discussed with the Consultant in Charge or on-call Consultant Haematologist
Product: Fibrogammin 250/1250 IU
Dosage
The dosing regimen should be individualised based on body weight, laboratory values, and the patient's clinical condition.
Routine prophylaxis dosing schedule for treatment of congenital FXIII deficiency
Prophylaxis should start with FXIII concentrate 20–40 iu/kg every 28 d, adjusted to maintain trough FXIII activity 0.1–0.2 iu/ml or 10-20 u/dl.
- Recommended dosing adjustments of ± 5 IU per kg should be based on trough FXIII activity levels as shown in Table 1 and the patient's clinical condition.
- Dosing adjustments should be made on the basis of a specific, sensitive assay used to determine FXIII levels.
Table 1: Dose adjustment
|
Factor XIII Activity Trough Level (%) |
Dosage Change |
|
One trough level of <10% |
Increase by 5 units per kg |
|
Trough level of 10% to 20% |
No change |
|
Two trough levels of >20% |
Decrease by 5 units per kg |
|
One trough level of >25% |
Decrease by 5 units per kg |
Prophylaxis prior to surgery & treatment of bleeding.
For mild bleeding or minor surgery in FXIII deficiency consider tranexamic acid
For severe bleeding or major surgery in FXIII deficiency, consider additional FXIII concentrate 10–40 iu/kg depending on the interval since last prophylaxis and severity of bleeding
After the patient's last routine prophylactic dose, if a surgery is scheduled:
- Between 21 and 28 days later – administer the patient's full prophylaxis dose immediately prior to surgery and the next prophylactic dose should be given 28 days later.
- Between 8 and 21 days later – an additional dose (full or partial) may be administered prior to surgery. The dose should be guided by the patient's FXIII activity levels and clinical condition and should be adjusted according to the half-life of Fibrogammin.
- Within 7 days of last dose – additional dosing may not be needed.
Options for treatment include tranexamic acid and rFVIIa
Recombinant FVIIa (Novoseven):
Dose, dose range and dose interval
The recommended dose range in adults and children for treatment of bleeding episodes and for the prevention of bleeding in patients undergoing surgery or invasive procedures is 15 – 30 µg per kg body weight every 4 – 6 hours until haemostasis is achieved. Dose and frequency of injections should be adapted to each individual.
1mg 2mg 5mg vials available
- Doses for FVII deficient patients should be prescribed as per dosing recommendations and NOT rounded to nearest whole vial size.
- Novoseven can be kept refrigerated following re-constitution and be used for multi dosing from a single vial for 24 hours.
- Novoseven is given by slow intravenous bolus injection either via butterfly, cannula or port-a-cath.
Following severe bleeds/injuries/some surgical interventions FVII monitoring may be undertaken. However, note the PT and FVII activity assays do not directly reflect the haemostatic activity of rFVIIa and have limited value in monitoring treatment. The plasma half-life of rFVIIa has been estimated as 2-8 h
Products – Tranexamic acid; FXI concentrate; SD treated FFP (Octaplas).
The bleeding phenotype in FXI deficiency is variable and does not always correlate with FXI activity. Some families have a significant bleeding tendency (higher bleeding risk cases) whereas others may be relatively asymptomatic.
FXI concentrate has been associated with a risk of thrombosis in adult patients.
Treatment
For minor bleeds or minor surgery in higher bleeding risk cases, and for all bleeds or surgery in low bleeding risk cases, consider tranexamic acid for 5–7 days.
For severe bleeds or major surgery in high bleeding risk cases, consider an initial dose of FXI concentrate 10–15 iu/kg, without additional tranexamic acid.
A combination of SD-FFP 15–25 ml/kg and tranexamic acid is an alternative to FXI concentrate
Haemarthrosis
- FVIII replacement may be required 12 hourly in a major joint bleed for first 24-48 hrs.
- Rest/elevate, where possible, the affected joint.
- Joint bleeds can be painful, analgesia should be advised/prescribed (aspirin containing products and NSAID's should not be prescribed)
- Physio referral for assessment should be made via Trakcare
Head Bumps/Knocks/Head Injuries
- Often relatively minor with small bumps/swelling on the head/forehead with some bruising; may require 1-2 doses of factor replacement. Management may require discussion with the on call haematologist.
- Children should be admitted for observation if the degree of trauma is significant or uncertain.
- Skull x-rays, CT scans etc. are not routinely required for minor injuries, but should be arranged promptly in the event of significant trauma or in the presence of symptoms or signs suggestive of intracranial bleeding.
- Factor levels should be monitored following major head trauma.
- Head injury guidelines are available in the haemophilia room or A&E dept to give to parents/carers, for those not requiring admission.
- Small scalp lacerations are usually best sutured or steri-striped, along with a dose of the appropriate factor product +/- tranexamic acid for 3-5 days. This can be carried out in A&E following appropriate factor replacement.
Mouth Bleeds
- Torn frenulum, bites/cuts to the tongue, can result in the loss of significant amount of blood.
- Those presenting with an oral bleed >4 hours should have haemoglobin checked.
- In addition to factor replacement a prescription for Tranexamic acid should be given for 3-5 days for all mouth bleeds for all patient groups.
- Ensure parents/carers are advised to complete the treatment programme prescribed even if the bleeding has stopped.
Musculoskeletal Bleeds
- Factor replacement is usually required daily for 24-48 hrs.
- Factor replacement may be required 12 hourly in a major muscle bleed, e.g. Iliopsoas bleed for 24-48hrs, followed by daily treatment for further 2-3 days.
Soft Tissue Bleeds/Injuries
- Tranexamic acid or factor replacement 1-2 days is usually sufficient.
Bleeding at Other Sites
- Deep cuts/lacerations should be treated as for musculo skeletal bleeds. If suturing is required they should be referred to A&E dept. or surgeons as appropriate following factor replacement.
- Fractures will require high dose factor replacement prior to any surgical management of the fracture by the orthopaedic team.
Review of Injuries/Bleeds
- Minor injuries/bleeds do not need to be reviewed the following day if parents/carers are happy that previous similar bleeding episodes have settled with single dose injections.
- Advate (rFVIII)
- RefactoAF (rFVIII)
- NovoEight (rFVIII)
- Elocta (extended half life rFVIII)
- Esperoct (extended half life FVIII)
- Novoseven (rFVIIa)
- Benefix (rFIX)
- Alprolix (extended half life rFIX)
- Idelvion (extended half life rFIX)
- Voncento (pd FVIII/vWF)
- DDAVP (IV/SC) Ward Fridges/pharmacy
- Tranexamic Acid Ward level/pharmacy
Reconstituting Factor Products
Most factor replacement products are packaged in 4 sizes:
250 IU 500 IU 1000IU 2000IU
- Doses are normally prescribed to allow complete vials to be administered.
- Differing batch numbers and vials sizes can be mixed in the same syringe.
- Reconstituted factor products should be given within three hours of reconstitution, unless a continuous infusion is being used.
- Factor products are given by slow intravenous bolus injection either via butterfly, cannula or port-a-cath.
- Factor products are obtained from blood bank, via the Trakcare system.
Please follow individual product insert/instructions for specific re-constitution guidance
For further information contact:
Medical staff (day time only):
|
Consultants: |
Dr E Chalmers |
85644 |
|
Haematology Registrar: |
Page/deg phone |
18437 |
Out of hours:
|
On call consultant or registrar |
(as per rota) |
Nursing Staff (day time only):
|
Advanced Nurse Practitioner |
Lyn Docherty |
84701 |
|
Haematology Nurse Specialist: |
Ruth Bissell |
84474 |
For Patient Information on Portal/Trakcare - see under care plans:
- Factor deficiency and level
- Treatment plan
Laboratory monitoring in patients on emicizumab:
Emicizumab affects routine standard bloods tests used to monitor FVIII replacement and tests used to assess inhibitors. Specific tests are therefore required.
For monitoring FVIII replacement in the presence of emicizumab use a chromogenic FVIII assay (Track request – see below). Note this assay is performed at GRI.
For measuring inhibitor levels request a chromogenic inhibitor assay (Track request – see below). Note this assay is performed at GRI.
On routine coagulation screening in patients on emicizumab the APTT should be shortened (or normal). Prolongation of the APTT may suggest the presence of an anti-drug antibody (ADA) against emicizumab. As this may affect efficacy further assessment will be required either using an emizicumab assay or a specific anti-drug antibody assay. This should be discussed with a consultant and/or the haemostasis lab.
Track requests for FVIII & inhibitor assays:
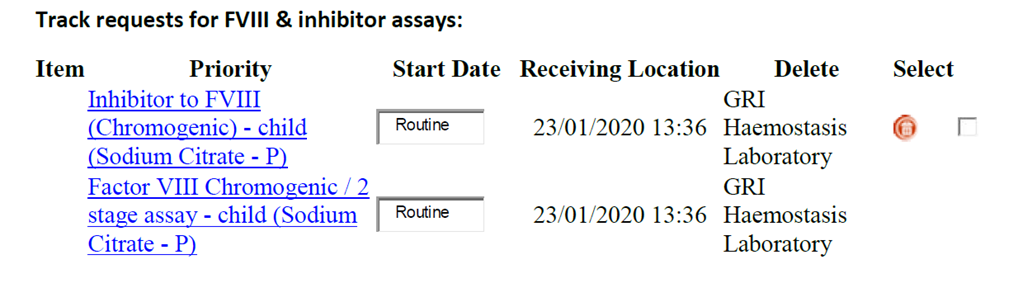
Additional information on treatment and monitoring is available on the UKHDCO website: http://www.ukhcdo.org/guidelines/
The following maintenance dose table is advisory. Some of the recommended doses represent a divergence from the licensed dose schedule. Dose-rounding has been applied at a margin of -/+ 10% of the calculated dose, which is felt to reflect better prescribing in practice.
The following presentations of Hemlibra® (Roche) are available:
|
Vial Quantity (mg) |
30 |
60 |
105 |
150 |
|
Vial Volume (ml) |
1.00 |
0.40 |
0.70 |
1.00 |
|
Vial Concentration (mg/ml) |
30 |
150 |
150 |
150 |
- The maximum permitted volume per single injection is 2.0ml. Doses which require a greater volume must be administered in separate injections each with a maximum volume of 2.0ml and must be injected at different sites.
- It is important to note that there is very limited data on the PK and PD in the very young and close monitoring is important.
Loading dose:
The loading dose is 3.0 mg/Kg weekly for 4 weeks (week 1-4) - see www.medicines.org.uk
Maintenance dosing
The following maintenance dose table has been devised with multiple aims:
- Risk minimisation through a preferential selection of less complex regimens
- Increased patient convenience by minimising the number of injections for each dose
- Two weekly regimens are the default recommendation for maintenance doses as these currently provide the best balance between patient safety, convenience and waste-minimisation but once-weekly regimens may be appropriate in the older group if they do not incur any additional wastage
- If only a partial quantity is required from any vial then this should be withdrawn from the vial with the smallest overall quantity with any remaining product being discarded
- It is good practice to avoid drawing up vials of different concentration into one syringe- 2 injections are required
- The dosing table will be updated periodically to reflect emerging evidence to support other regimens which offer additional or comparable patient convenience, safety and waste-minimisation and when there is more clinical experience or evidence emerges for doses >150mg weekly (or equivalent)
Additional Factor VIII:
- Patients will require a small supply of factor VIII to administer should a bleed occur
- A small quantity of a recombinant factor VIII with a low acquisition cost should be supplied to the patient
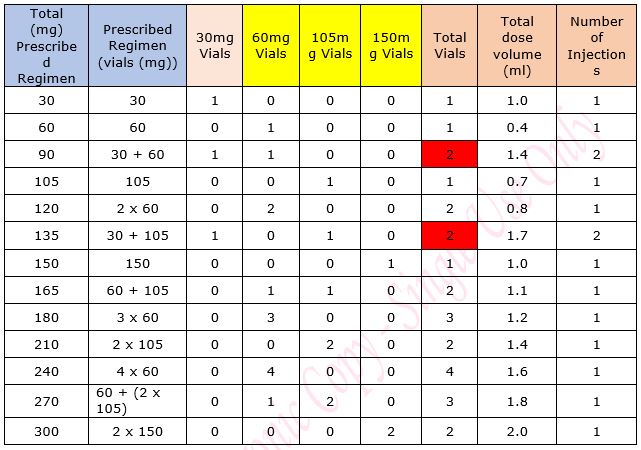
Table 1: Maintenance Dose Table for Emicizumab for Haemophilia A in patients without an inhibitor
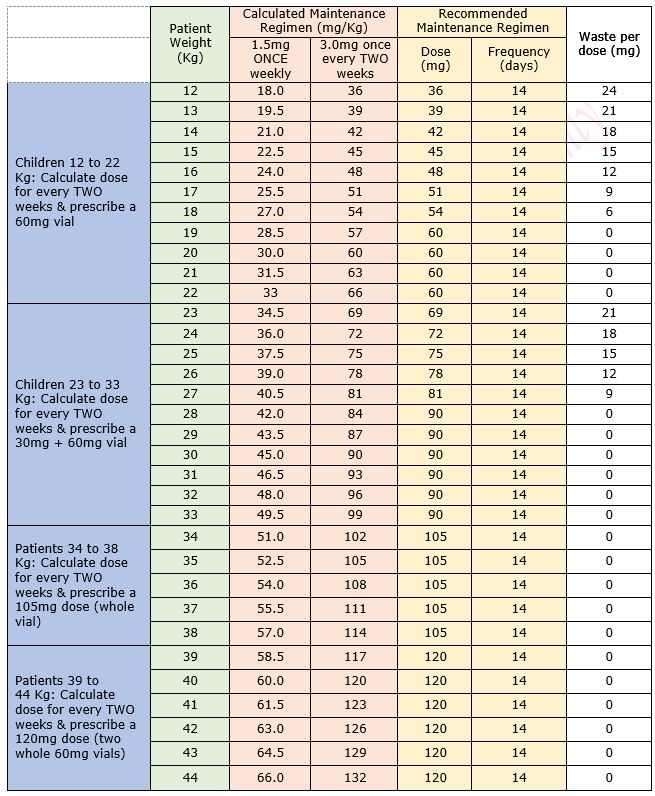
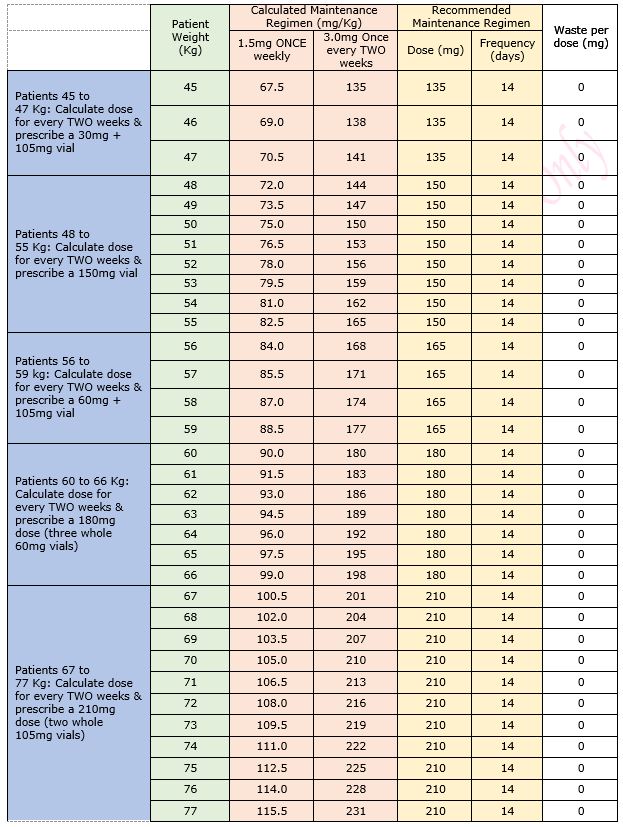
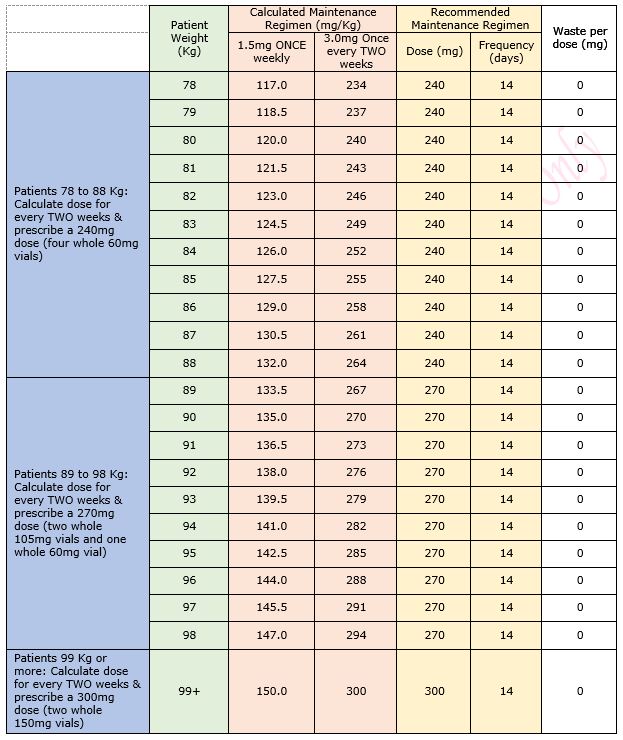
Table 2: Regimen details
- Select patient from Trakcare
- Know which product you need (see care plan)
- Select NEW REQUEST
- Select Labs Child
- Tick Specimen Collection Box
- Sub Category- Delete VIROLOGY SPEC and enter BT in box, click on spy glass
- Select Blood Transfusion
- Item Box - enter OT, click on spy glass
- Select Other Products
- Add to list
- PRIORITY BOX- hit spy glass Select URGENT
- Update
- Complete the boxes, mandatory where question in bold
- Products required- enter name of product and how many units/vials needed, bearing in mind vial sizes
- Enter the location that you want factor delivered to
- Complete with personnel password.
- The TrakCare request form will print in the lab but you will also have to alert the lab by phone and send a collection card (see Appendix 4)
- If pod system is ‘down’ use emergency porter.
- All requests to treat a bleeding episode/injury should be treated as urgent.
- To have order delivered YOU MUST page the MLA pg 17262, to request delivery of the product, (you will need the patient CHI number to confirm identity).
If the Blood Product Collection Card is unable to be printed in blood bank then this will have to be completed and sent to the lab.

Diagnosis and treatment of factor VIII and IX inhibitors in congenital haemophilia: (4th edition). Peter W Collins, Elizabeth Chalmers (et al). British Journal of Haematology (BJH), 2013, 160, 153-170
Guideline for the diagnosis and management of the rare coagulation disorders. A United Kingdom Haemophilia Centre Doctors’ Organisation guideline on behalf of the British Committee for Standards in Haematology. Andrew D Mumford, Writing Group Chair & BCSH Task Force Member, Sam Ackroyd, Raza Alikhan, Louise Bowles, Pratima Chowdary, John Grainger, Jason Mainwaring, Mary Mathias and Niamh O’Connell on behalf of the BCSH Committee. BJH 2014, 167, 304-3326
The diagnosis and management of von Willebrand disease: a United Kingdom Haemophilia Centre Doctors’ Organisation guideline approved by the British Committee for Standards in Haematology. Mike A Laffan, Will Lester, James S O’Donnell, Andrew Will, Robert Campbell Tait, Anne Goodeve, Carolyn M Millar and David M Keeling. BJH 2014, 167, 453-465
The use of enhanced half-life coagulation factor concentrates in routine clinical practice: guidance from UKHCDO. P Collins, E Chalmers (et al). Haemophilia 2016, 22, 487-498
Treatment of bleeding episodes in haemophilia A complicated by a factor VIII inhibitor in patients receiving Emicizumab. P. W. Collins, R. Liesner, M. Makris, K. Talks, P. Chowdary, E. Chalmers, G. Hall, A. Riddell C. L. Percy, C. R. Hay, D. P. Hart Haemophilia 2018
Last reviewed: 30 November 2020
Next review: 30 November 2022
Author(s): Dr E Chalmers
Version: 6
Approved By: Schiehallion Clinical Governance Group
Document Id: RHC-HAEM-ONC-006